Lesson 1: What is Biowulf?
To fully engage with the course material and complete the hands-on exercises, we'll be leveraging the powerful NIH HPC Biowulf system.
To make the most of this powerful tool, it's essential to grasp the fundamentals of working with HPC systems, specifically Biowulf. In this lesson and others in Module 1, we will delve into the key concepts and practical skills you'll need to navigate this environment effectively.
Learning Objectives
- Understand the components of an HPC system. How does this compare to your local desktop?
- Learn about Biowulf, the NIH HPC cluster.
- Learn about the command line interface and resources for learning.
Additional training materials at hpc.nih.gov
Much of the content for this presentation is from hpc.nih.gov. For more information and more detailed training documentation, see hpc.nih.gov/training/.
Note
This lesson will be demo-based only. Hands-on lessons will not begin until Lesson 2.
What is a high performance cluster (HPC)?
A collection of standalone computers that are networked together. They will frequently have software installed that allow the coordinated running of other software across all of these computers. This allows these networked computers to work together to accomplish computing tasks faster. --- hpc-intro (Software carpentries)
NIH HPC Systems
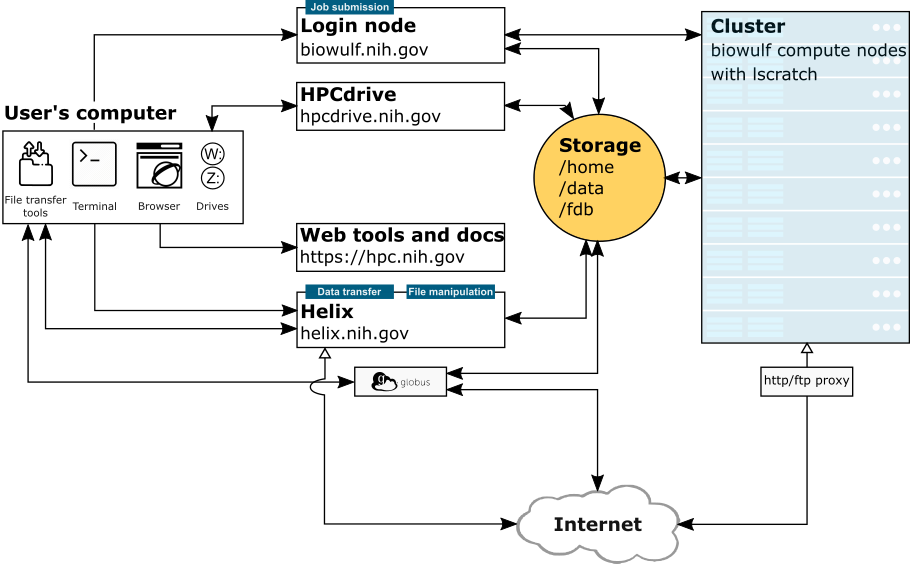
Image Credit: hpc.nih.gov/systems
This diagram describes the NIH HPC Systems (Login node, Biowulf Cluster, HPC drive, and Helix), how the systems interact, and how you interact with the systems. Each "system" is described here. We will also discuss these further below.
When using an HPC
-
We use a command line interface and a Secure shell protocol (SSH) to establish a remote connection to the login node / head node.
-
Most of us are likely used to a graphical user interface (GUI), which is a point-and-click interface, meaning we use a mouse to move around and click on various display icons. We visually interact with our compute environment. However, we do not generally interact with an HPC in this way. HPCs are remote resources that require connections using slow or intermittent interfaces (over WIFI and VPNs). Because of this, it is more practical to guide functionality over the command line using plain text.
-
The cluster head node (login node) distributes compute tasks (the things we want to do outside of file manipulation and editing) using a scheduling system (e.g., SLURM). We use a scheduling system because the HPC is a shared resource with hundreds of worker nodes and thousands of processors.
-
Note
Slurm stands for Simple Linux Utility for Resource Management.
What is Biowulf?
Biowulf is the National Institutes of Health's (NIH) high-performance computing (HPC) cluster. This massive Linux system boasts over 90,000 processors, allowing it to tackle enormous computational tasks by dividing jobs and running them simultaneously across multiple nodes. Biowulf comes pre-loaded with over 600 scientific software programs and databases relevant to various fields like genomics, molecular biology, and bioinformatics. For security reasons, access to Biowulf is restricted to the NIH campus network or through a Virtual Private Network (VPN).
When should we use Biowulf?
You should use Biowulf when:
- Software is unavailable or difficult to install on your local computer and is available on Biowulf.
- You are working with large amounts of data that can be parallelized to shorten computational time AND/OR
- You are performing computational tasks that are memory intensive.
Example of High Performance Computing Structure

Essentially Biowulf is a scaled up version of your local computer.
In Biowulf, many computers make up a cluster. Each individual computer or node has disk space for storage and random access memory (RAM) for running tasks. The individual computer is composed of processors, which are further divided into cores, and cores are divided into CPUs.
Info
Information on the NIH HPC architecture and hardware here.
Getting an NIH HPC account
- If you do not already have a Biowulf account, you can obtain one by following the instructions here.
- NIH HPC accounts are available to all NIH employees and contractors listed in the NIH Enterprise Directory.
- Obtaining an account requires PI approval and a nominal fee of $40 per month.
- Accounts are renewed annually contigent upon PI approval.
Node types on the HPC
- Login node (head node)
- Used for submitting resource intensive tasks as jobs
- Editing and compiling code
- File management and data transfers on a small scale
- Compute nodes (worker nodes)
- For computational processes
- Requires interaction with a job scheduling system (SLURM)
- Batch jobs, sinteractive sessions
- Data transfer node (For Biowulf, this is Helix.)
Info
sinteractive - work on biowulf compute nodes interactively; suitable for testing/debugging cpu-intensive code, Pre/post-processing of data, and using graphical applications.
sbatch - for submitting shell scripts via jobs, taking away any interactive component.
swarm - used for runnning embarassingly parallel code as independent jobs.
The Data transfer node: Helix
- Used for data transfers and file management on a large scale.
- 48 core system with 1.5 TB of main memory
- direct internet connection
- Helix should be used when
- you are transferring >100 GB using
scp - gzipping a directory containing >5K files, or > 50 GB
- copying > 150 GB of data from one directory to another.
- uploading or downloading data from the cloud.
- you are transferring >100 GB using
- For more information on data transfers see hpc.nih.gov.
Biowulf Data Storage

- You may request more space on
/data, but this requires a legitimate justification. - More information on data storage here.
Important
Data storage on the HPC system should not be for archival purposes.
Note
Though there aren't true back-ups of your data directories, there are snapshots with a view of your home and data directories at a specific point in time. You can learn more about snapshots in the HPC documentation.
-
To check disk space use:
checkquota- this shows the directories for which you have write accessOR
Look on the *user dashboard -> disk storage.
*Only works on VPN
Best practices file storage

How do I create a directory in scratch?
mkdir /scratch/$USER
Applications on Biowulf
- Bioinformatics applications and other programs are available on Biowulf via modules.
- View a list of available applications here.
Info
Loading software as environment modules allows us to better control our computational environment and easily use a large number of programs and even different versions of the same programs. Modules alter the user's environment variables such as the execution path.
The Command Line Interface (CLI)
What is Unix?
- Unix is a proprietary operating system like Windows or MacOS (Unix based).
- There are many Unix and Unix-like operating systems, including open source Linux and its multiple distributions.
- Biowulf nodes use a Unix-like (Linux) operating system (distributions RHEL8/Rocky8).
- Biowulf requires knowledge and use of the command line interface (shell) to direct computational functionality.
- To work on the command line we need to be able to issue Unix commands to tell the computer what we want it to do.
Tip
A basic foundation of Unix is advantageous for most scientists, as many bioinformatics open-source tools are available or accessible by command line on Unix-like systems.
Accessing your local terminal or command prompt
Mac OS
Type cmd + spacebar and search for "terminal". Once open, right click on the app logo in the dock. Select Options and Keep in Dock.
Windows 10 or greater
You can start an SSH session in your command prompt by executing ssh user@machine and you will be prompted to enter your password. ---Windows documentation
To find the Command Prompt, type cmd in the search box (lower left), then press Enter to open the highlighted Command Prompt shortcut.
Are you a Windows user and not affiliated with NIH?
If you are using a Windows operating system, Windows 10 or greater, you can use the Windows Subsystem for Linux (WSL) for your computational needs.
The Windows Subsystem for Linux (WSL) is a feature of the Windows operating system that enables you to run a Linux file system, along with Linux command-line tools and GUI apps, directly on Windows, alongside your traditional Windows desktop and apps. --- docs.microsoft.com
To install WSL, follow the instructions here. There are multiple Linux distributions. We recommend new users install "Ubuntu".
Windows WSL is not available to NIH employees due to security policies.
How much Unix do we need to learn?
As with any language, the learning curve for Unix can be quite steep. However, to work on Biowulf you really need to understand the following:
- Navigating the File System: Understanding the hierarchical structure of directories, using the
cdcommand to move between directories. - File Paths: Learning how to specify the location of files using absolute and relative paths.
- Basic Unix Commands: Getting acquainted with common commands like
lsfor listing files,mvfor moving files,rmfor removing files,mkdirfor creating directories,catfor viewing file contents, andmanfor accessing command documentation. - Getting help: Discovering how to find more information about Unix commands and their usage.
- Command Customization: Learning how to modify the behavior of Unix commands using flags or options, such as using
ls -lto list files with detailed information compared to the basiclscommand. - Redirecting Input and Output: Understanding standard input and output, and how to redirect the output of one command to the input of another using pipes (
|) or redirection operators (>,<).
More on these in the next lesson.
Connecting to Biowulf
-
To connect to Biowulf, we use a secure shell (SSH) protocol.
Establishing a remote connection
"username" = NIH/Biowulf login username.
Note
If this is your first time logging into Biowulf, you will see a warning statement with a yes/no choice. Type "yes".
Type in your password at the prompt. The cursor will not move as you type your password!
HPC OnDemand
Recently, the NIH HPC Team has provided on demand access to HPC resources via web browser through integration of Open OnDemand. This integration makes working with HPC resources less intimidating for new users, as they will not have to open a terminal and remotely connect via ssh. Instead, navigate to your web browser (Google Chrome is preferred) and connect to NIH HPC OnDemand using https://hpcondemand.nih.gov/.
HPC OnDemand provides an online dashboard for users to easily access command line interactive sessions, graphical linux desktop environments, and interactive applications including RStudio, MATLAB, IGV, iDEP, VS Code, and Juptyer Notebook.

HPC OnDemand Dashboard
SLURM commands
You will also need to know commands specific to the Biowulf job scheduling system:
sbatchsubmit slurm jobswarmsubmit a swarm of commands to clustersinteractiveallocate an interactive sessionsjobsshow brief summary of queued and running jobssqueuedisplay status of slurm batch jobscanceldelete slurm jobs
We will talk about many of these in more detail in later lessons.
How to load / unload a module
-
To see a list of available software in modules use
-
To load a module
-
To see loaded modules
-
To unload modules
Note
You may also create and use your own modules.
Getting help on Biowulf: NIH HPC Documentation
The NIH HPC systems are well-documented at hpc.nih.gov.
Note
Existing safeguards make it nearly impossible for individual Biowulf users to irreparably mess up the system for others.
WORST CASE SCENARIO - You are locked out of your account pending consultation with NIH HPC staff
Additional HPC help
-
Contact staff@hpc.nih.gov
The HPC team welcomes questions and is happy to offer guidance to address your concerns. -
Monthly Zoom consult sessions
The HPC team offers monthly zoom consult sessions. "All problems and concerns are welcome, from scripting problems to node allocation, to strategies for a particular project, to anything that is affecting your use of the HPC systems. The Zoom details are emailed to all Biowulf users the week of the consult." -
Bioinformatics Training and Education Program
If you experience any difficulties or challenges, especially with different bioinformatics applications, please do not hesitate to email us at BTEP.
User Dashboard

- Can view disk usage and job info
- Request more disk space
- Evaluate job info for troubleshooting
Learning Unix: Classes / Courses
- Introduction to Biowulf (May – Jun, 2023)
- Introduction to Unix on Biowulf (Jan – Feb, 2023)
- Bioinformatics for Beginners: Module 1 Unix/Biowulf
Additional Unix Resources:
Key points
- Biowulf is the high performance computing cluster at NIH.
- To work on Biowulf, you will need to use the command line interface, which requires some knowledge of unix commands.
- When you apply for a Biowulf account you will be issued two primary storage spaces:
/home/$User(16 GB)/data/$USER(100 GB).
- Hundreds of pre-installed bioinformatics programs are available through the
modulesystem. - Computational tasks on Biowulf should be submitted as a job (
sbatch,swarm) or through an interactive session (sinteractive). - Do not run computational tasks on the login node.