Lesson 1: Introduction to Unix and the Shell
Lesson Objectives
-
Course overview.
-
Introduce Unix and describe how it differs from other operating systems.
-
Introduce and get set up on DNAnexus and the GOLD system.
-
Discuss ways to use the command line outside of the DNAnexus teaching environment.
-
Introduce conda.
Why Bioinformatics?
-
Analyze your own data
-
Expand scientific training and skills
-
Provide a path to a new career
-
Have a better understanding of how other people analyze data
Useful concepts / guidance from statistics
Terms to know
-
Normalization - the process of scaling data to account for uncontrolled factors affecting variation.
-
Effect size - "the quantitative measure of the magnitude of a phenomenon" (Biostar Handbook).
-
P-value - "the probability for the observed effect size to be a product of random chance" (Biostar Handbook). Statitistical significance is usually set to values below 0.05 or 0.01.
-
Adjusted p-value adjusted for multiple testing / multiple comparisons. When repeating a test multiple times, the chances of getting a value by random chance increases.
-
Statistical power - "reflects the ability of a test to produce the 'correct' prediction" (Biostar Handbook). This is impacted by sample size, effect size, and the applied statistical model.
-
Confidence interval - the range of values that contains some true value at a defined probability (e.g., 95%).
Additional guidance
-
Statistical Models - these should be appropriate for your experimental design. The methods you are using should be consistent with what's in related scientific literature. Different tests produce different results. Consult a statistician if possible.
-
Outliers - try to identify early on. If you remove data, you should have sound rationale for doing so.
-
P-hacking and HARKing - P-hacking is when you alter some form of the data analysis pipeline to get different results (e.g., using different statitical tests or including/excluding only subsets of the data). HARKing is modifying your hypotheses based on results. These are common in data science, and can be dangerous if (1) you aren't transparent about why something was done and what you did, (2) you fail to validate results, and (3) you don't explore alternative explanations.
Do not overinterpret the meaning of a p-value. p-value thresholds are fairly arbitrary.
What is Unix?
-
An operating system, just like Windows or MacOS
-
Something that is worth learning
-
Sometimes used interchangeably with Linux, which for our purposes, is just a version of Unix
Why learn Unix?
-
Many tools (like a bazillion) for biological data analysis are freely available and supported on Unix systems
-
Useful for working with big data, like genomic sequence files
-
To use the NIH High Performance Cluster (HPC) Biowulf for data analysis
A few things about the Unix shell...
-
It gives a command line interface where users can type commands
-
Also a scripting language, used to automate repetitive tasks
-
The Bash shell (the Bourne Again SHell) is the most popular Unix shell.
How is Unix different from other operating systems?
-
Does not use a Graphical User Interface (GUI) better known as a "point and click" environment.
-
The user has to learn a series of commands for interacting with a Unix system
-
BUT...a few commands, like the ones we will learn over the next several lessons, will allow us to employ a number of bioinformatics tasks
How much Unix do I need to know to get started?
As with any language, the learning curve for Unix can be quite steep. However, to get started analyzing data you really need to understand the following:
- Directory navigation: what the directory tree is, how to navigate and move around with cd
- Absolute and relative paths: how to access files located in directories
- What simple Unix commands do: ls, mv, rm, mkdir, cat, man
- Getting help: how to find out more on what a unix command does
- What are “flags”: how to customize typical unix programs ls vs ls -l
- Shell redirection: what is the standard input and output, how to “pipe” or redirect the output of one program into the input of the other --- Biostar Handbook
Many of these will be covered in Lesson 2.
Getting started with DNAnexus
What is DNAnexus?
DNAnexus provides a secure cloud based platform for the analysis and sharing of next generation sequencing data. This class will use a pre-built teaching environment, the GOLD platform, which includes all of the software needed installed and ready to go.
Obtaining a DNAnexus account
If you have not already created a free DNAnexus account, please do so here. Once you have obtained your free account, you will need to email us your username at ncibtep@nih.gov to obtain access to the course page and GOLD System.
Finding the course and getting started with the GOLD system
Step 1: Login to DNAnexus
Step 2: Once you login, you should see the Projects page. If you have used DNAnexus previously, you may see more than one project listed. If this is your first time using DNAnexus, you will only see the project name for this course listed, BioStars. Double click on BioStars.

Step 3: Once you double click on the BioStars project, you will see a project directory containing multiple subdirectories and files. Select (double click) one of the .html files (e.g., Class_LETTER_LETTER.html). We have divided the class into four groups based on name. For example, if your first name begins with a letter A-E, select Class_A_E.html; if your first name begins with a letter F-M, select Class_F_M.html. You will need to double-click on the .html file.
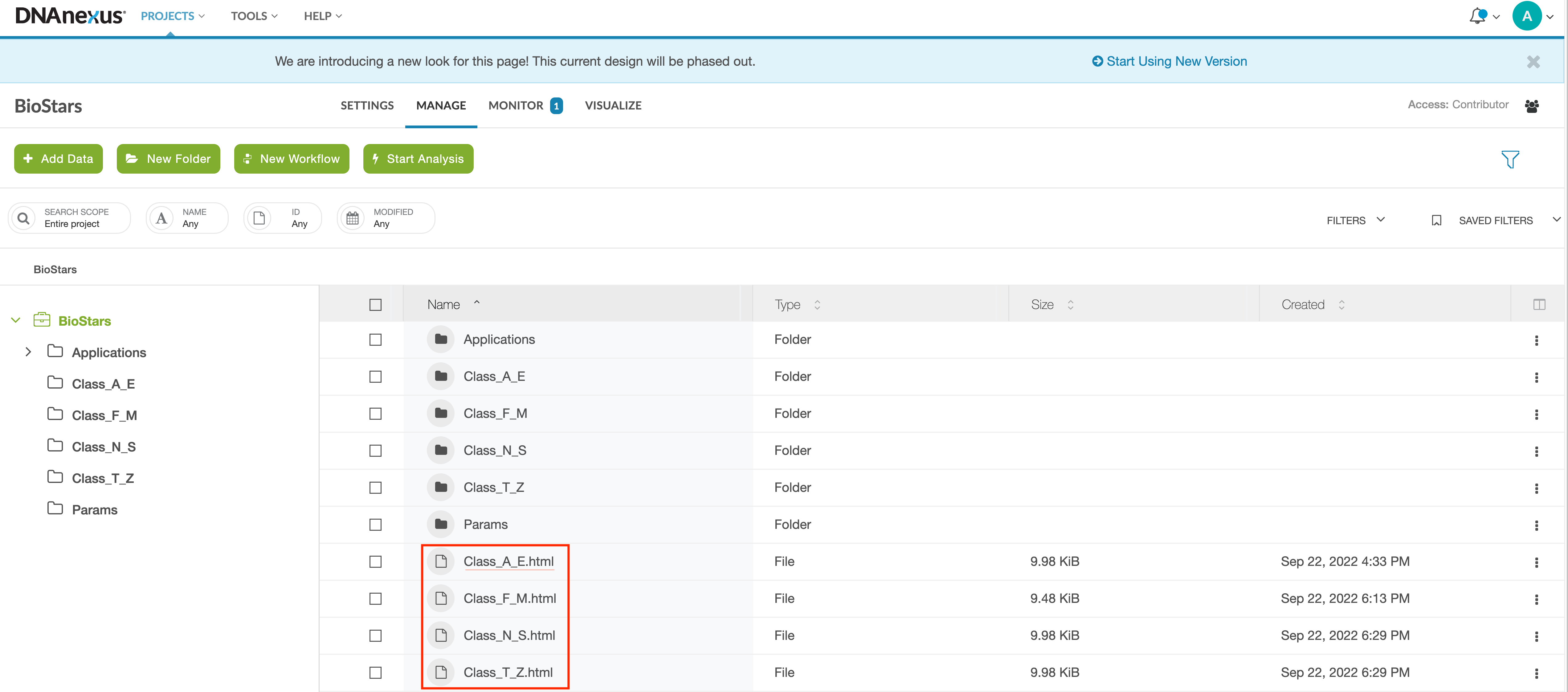
Step 4: The Class_LETTER_LETTER.html file will open the GOLD platform application, and you will see a screen that looks like this:
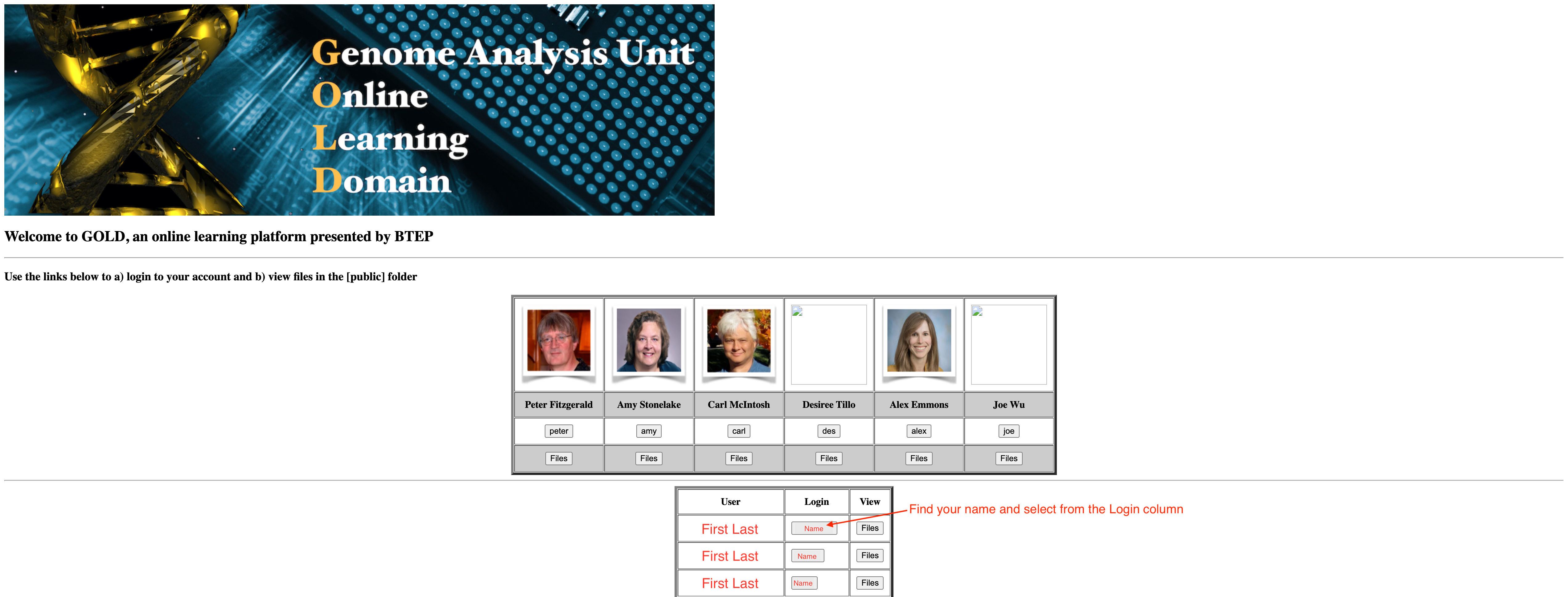
At the top of the page you will see the instructors pictures and logins. You will need to find your name (First and Last) in the table below the instructors. Once you find your name click on the link associated with your name in the login column. The name that you see in the login column will serve as your username in step 5.
Step 5: The login link will open a terminal with a prompt to login. Login with your username (See step 4) and password (to be distributed in class).

Step 6: Once you login at the terminal, you will see the following page:
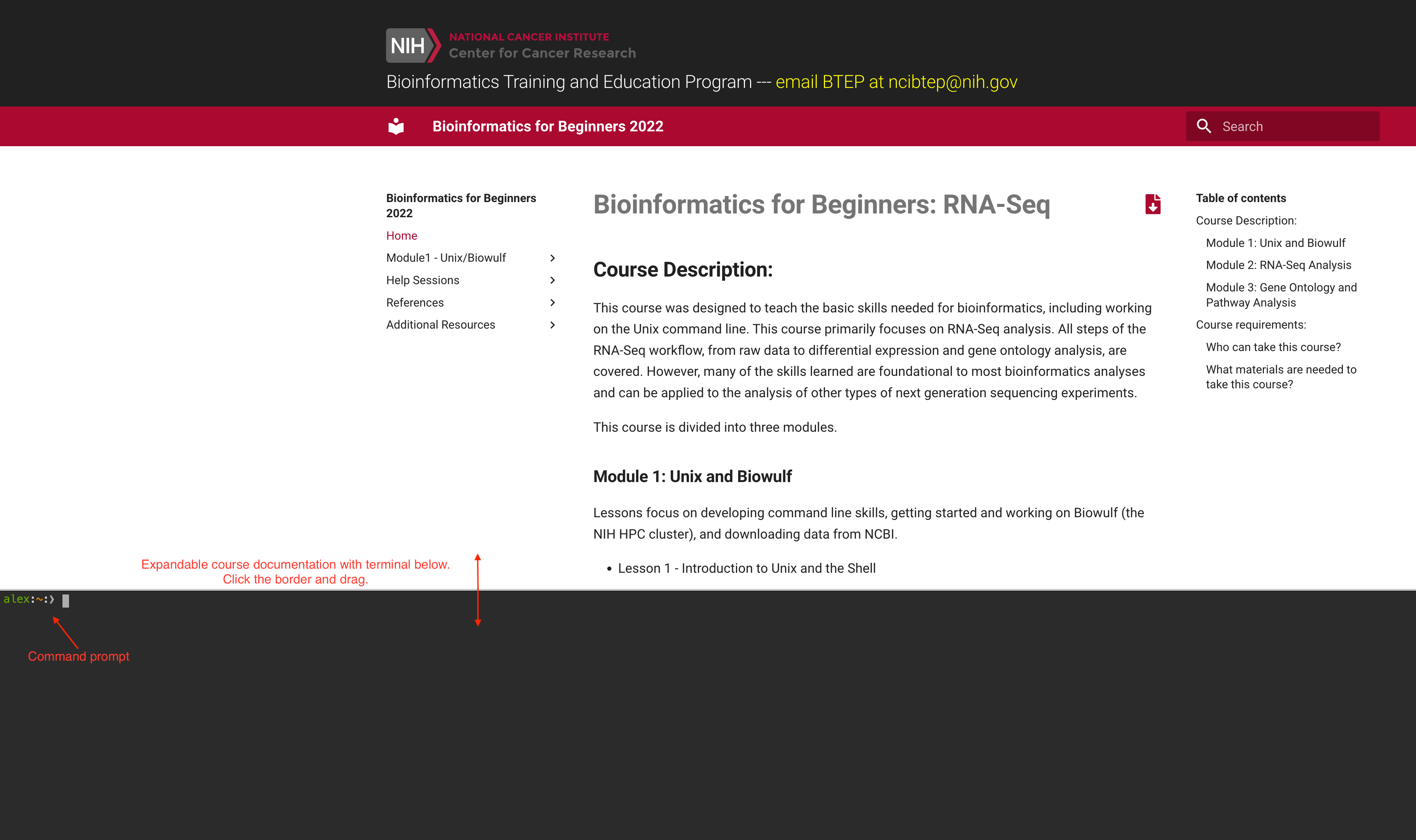
The course documentation is accessible at the top of the page and can be dragged up or down for viewing. The command line terminal accounts for the rest of the page. You may need to resize the screen to see the command prompt.
Now you should be logged onto the GOLD platform and ready for class.
Ending your DNAnexus session: if you are finished with the GOLDsystem for the day, logout using
exit
Getting started outside of DNAnexus
Lessons 3 and 5 will focus on Biowulf and will require VPN access and the ability to connect remotely via a terminal on your local computer.
In addition, we want you to be able to get started analyzing data on your own without having to use the GOLD teaching environment. Most bioinformatics software will work with unix based systems (MacOS or Linux). Therefore, if you are working on a Windows operating system, you will need a work around.
Working with command line from a macbook
-
Type
cmd + spacebarand search for "terminal". Once open, right click on the app logo in the dock. SelectOptionsandKeep in Dock. -
The default shell starting with Mac OSX version 10.14 is the
zshshell. While this is not really a problem, you can configure your computer to use the bash shell using the following:
chsh -s /bin/bash
- Follow Biostar instructions if you want
bioinfo, which contains the software used in the book, installed on your local machine. Regardless, you will likely need to install the Xcode compiler. You can search for and install this directly from the "App Store". Then install the additional Xcode command line tools from the terminal using
xcode-select --install
Working with command line from a Windows computer
If you are using a Windows operating system, Windows 10 or greater, you can use the Windows Subsystem for Linux (WSL) for your computational needs.
The Windows Subsystem for Linux (WSL) is a feature of the Windows operating system that enables you to run a Linux file system, along with Linux command-line tools and GUI apps, directly on Windows, alongside your traditional Windows desktop and apps. --- docs.microsoft.com
To install WSL, you will need to submit a help ticket to service.cancer.gov. There are multiple Linux distributions. We recommend new users install "Ubuntu".
If you do not plan to use your local machine for bioinformatics analyses, you can connect to the NIH HPC Biowulf using an SSH client. The secure shell (ssh) protocol is commonly used to connect to remote servers. More on Biowulf later.
Windows 10 or greater has a built in SSH client.
You can start an SSH session in your command prompt by executing ssh user@machine and you will be prompted to enter your password. ---Windows documentation
To find the Command Prompt, type cmd in the search box (lower left), then press Enter to open the highlighted Command Prompt shortcut.
If this yields any major issues, try installing PuTTY, Solar-PuTTY, or MobaXterm.
Note about command prompt and powershell: Just like the bash shell works effectively with a linux operating system, Windows also has shells to interact with the Windows operating system. Windows has two shells: the Command Prompt and the PowerShell. However, because most bioinformatics software is unix based, these shells will not be useful for bioinformatics scripting.
Working with the Biostars software on Biowulf
For your convenience, we have created a module on Biowulf that includes many of the same programs in the bioinfo environment from The Biostar Handbook. To use this module, please see the instructions documented under Additional Resources.
What is conda?
The Biostar Handbook works with programs installed within a conda environment named bioinfo. Conda is commonly used for bioinformatics package installations.
Conda is often used for scientific software installation because...
Installing software is hard. Installing scientific software is often even more challenging. In order to minimize the burden of installing and updating software (data) scientists often install software packages that they need for their various projects system-wide.
Installing software system-wide has a number of drawbacks:
It can be difficult to figure out what software is required for any particular research project. It is often impossible to install different versions of the same software package at the same time. Updating software required for one project can often “break” the software installed for another project. --- Pugh and Tocknell, Introduction to Conda for (Data) Scientists
Conda solves these problems by facilitating software installations, making the installation process far easier. As a package and environment management system, conda also enhances both the portability and reproducibility of scientific workflows by isolating software and their dependencies in "environments". These environments do not interact with system wide programs and therefore do not reek havoc on your local machine due to software incompatibilites.
Conda runs on Windows, macOS, Linux and z/OS. Conda quickly installs, runs and updates packages and their dependencies. Conda easily creates, saves, loads and switches between environments on your local computer. It was created for Python programs, but it can package and distribute software for any language. --- docs.conda.io
Activating / deactivating a conda environment
The Biostar handbook has included bioinformatics software in a conda environment named bioinfo. If you followed Biostar Handbook instructions and created a bioinfo environment on your local computer, you will need to activate the environment to use installed software.
To activate a conda environment use
conda activate bioinfo
To deactivate your environment
conda deactivate
The importance of documentation
Analyzing next generation sequencing data requires a large number of computational steps. As you work, you should ALWAYS keep a record of what you are doing. Just as you keep a laboratory notebook, you should have a notebook for bioinformatics, in which you record the programs that you used, including version information, what commands you entered and their parameters, and any other code that you used to prepare or move around data. Your collaborators and future self will thank you. There are many options out there for code editors; both Visual Studio Code and Atom are fairly popular and approachable for beginners. Consider keeping a README.txt log per project or analysis step.
Help Session
- Getting set up on DNAnexus
- Getting everyone access to Biowulf via
ssh.