Introduction to scRNA-Seq with R (Seurat)
This lesson provides an introduction to R in the context of single cell RNA-Seq analysis with Seurat.
Learning Objectives
- Learn about options for analyzing your scRNA-Seq data.
- Learn about resources for learning R programming.
- Learn how to import your data for working with R.
- Learn about Seurat and the Seurat object including how to create the object and access and manipulate stored data.
Exploratory Data Analysis: scRNA-Seq
This tutorial assumes that all pre-processing steps (read demultiplexing, FASTQ QC, reference based alignment, error correction) have been completed. At this stage of the analysis, a gene-by-cell count matrix has been generated for each sample.
Analysis Ecosystems
Following Cell Ranger and/or other pre-processing tools, you will have a gene-by-cell counts table for each sample. The three most popular frameworks for analyzing these count matrices include:
-
R (Seurat).
- Seurat, brought to you by the Satija lab, is a kind of one-stop shop for single cell transcriptomic analysis (scRNA-seq, multi-modal data, and spatial transcriptomics). It has a wide user base and is scalable, especially with Seurat v5. It is a great place to get started analyzing your data.
-
R (Bioconductor)
- If you are familiar with Bioconductor, a project and package repository for analyzing biological data, the Bioconductor ecosystem is a fantastic choice. This workflow will use many different, but highly interoperable packages. Orchestrating Single-Cell Analysis with Bioconductor is a fantastic resource for basic to more complicated types of analyses.
-
Python (scverse)
scverse, which is a multi-package ecosystem in python (core packages:scanpy,muon,scvi-tools,scirpy, andsquidpy), is the obvious choice for python users, and boasts advantages such as "scalability, extendability, and strong interoperability."
These options require some knowledge of either R or Python programming languages. Which one you choose will most likely depend on your preference, your PI's preference, and your goals. Here we will focus on the Seurat ecosystem.
Note
You may benefit by working with tools from all three of these ecosystems. This can be done by converting object types using a variety of packages (e.g. Seurat, sceasy, zellkonverter). However, there may be some hurdles; for example, the Seurat function as.SingleCellExperiment() does not seem to work with Seurat v5 layers. Therefore, hopping between systems may require some degree of web searching or creative workarounds.
Point-and-click alternatives
If you lack programming skills and learning to code seems daunting, there are GUI based alternatives including, but not limited to the following:
- Partek Flow (License available to NCI researchers)
-
Relevant trainings:
-
Qlucore Omics Explorer (License available to NCI-CCR researchers)
-
NIH Integrated Data Analysis Platform
What is R?
R is both a computational language and environment for statistical computing and graphics. It is open-source and widely used by scientists, not just bioinformaticians. Base packages of R are built into your initial installation, but R functionality is greatly improved by installing other packages.
R is a particularly great resource for statistical analyses, plotting, and report generating. The fact that it is widely used means that users do not need to reinvent the wheel. There is a package available for most types of analyses, and if users need help, it is only a Google search away. As of now, CRAN houses +20,000 available packages. There are also many field specific packages, including those useful in the -omics (genomics, transcriptomics, metabolomics, etc.). For example, the latest version of Bioconductor (v 3.18) includes 2,266 software packages, 429 experiment data packages, 920 annotation packages, 30 workflows, and 4 books.
What is RStudio?
RStudio is an integrated development environment for R, and now python. RStudio includes a console, editor, and tools for plotting, history, debugging, and work space management. It provides a graphic user interface for working with R, thereby making R more user friendly. RStudio is open-source and can be installed locally or used through a browser (RStudio Server or Posit Cloud).
What is Seurat?
Seurat is an R package designed for QC, analysis, and exploration of single-cell RNA-seq data. Seurat aims to enable users to identify and interpret sources of heterogeneity from single-cell transcriptomic measurements, and to integrate diverse types of single-cell data. ---https://satijalab.org/seurat/
Advantages:
- beginning-to-end workflows
- Nice vignettes
- Regularly updated
- Somewhat extensible
- Scalable
Disadvantages:
- updates break some functionality
- maintained by a single group
- Many functions are poorly documented
Updates with Seurat v5
Seurat v5 introduced the following new features:
- Integrative multi-modal analysis with bridge integration
- ‘Sketch’-based analysis of large data sets
- methods for spatial transcriptomics
- assay layers
You can read about major changes between Seurat v5 and v4 here.
How much R programming do I need to know to use Seurat?
The answer to this question will largely depend on the user. While some will be able to learn R on the go, others will need to know some amount of R programming prior to beginning their analysis. If needed, check out the quick R refresher under Things to know about R. There are also several R courses available through BTEP with self-contained, asynchronous content that can be used to learn independently of live sessions.
The most important thing to know is how to find help!
How to find help?
ALWAYS, ALWAYS, ALWAYS read the documentation.
Use the help pane in the lower right of RStudio or the functions, help() and help.search() or ? and ??.
Check out package vignettes (vignette()).
Check the Github site if one is available for help with a specific package. There may also be known issues listed under the Github Issues tab (Seurat issues).
Google for help!
Use Google to troubleshoot error message or simply find help performing a specific task. But, make sure you are precise and informative in your search. Stack Overflow is a particularly great resource. A good Google search includes 3 elements:
- The specific action (e.g., how to rename a column).
- The programming language (e.g., R programming).
- The specific style/technique for coding (e.g., dplyr or tidyverse package) Example: "How to rename a column in R with dplyr/tidyverse”. --- https://crd230.github.io/tips.html.
If this fails, feel free to email us at BTEP.
Things to know about R
R can be accessed from the command line using R, which opens the R console, or it can be accessed via and Integrated development environment (IDE) (e.g., RStudio, VSCode, etc.). R commands can be submitted together in a script or interactively in a console.
You can install and use R locally or via an HPC such as the NIH HPC Biowulf.
R is case sensitive, so avoid typos, and space agnostic.
How to navigate directories
setwd()Set working directory (equivalent tocd)getwd()Get working directory (equivalent topwd)
Getting help
-
help()and?"provide access to the documentation pages for R functions, data sets, and other objects". -
help.search()"allows for searching the help system for documentation matching a given character string in the (file) name, alias, title, concept or keyword entries (or any combination thereof)"; equivalent to??pattern
See more on getting help here.
Installing and loading packages
To take full advantage of R, you need to install R packages. R packages are loadable extensions that contain code, data, documentation, and tests in a standardized shareable format that can easily be installed by R users. The primary repository for R packages is the Comprehensive R Archive Network (CRAN). CRAN is a global network of servers that store identical versions of R code, packages, documentation, etc (cran.r-project.org).
An R library is, effectively, a directory of installed R packages which can be loaded and used within an R session. ---renv
install.packages()install packages from CRANlibrary()load packages in R sessionBiocManager::install()install packages from Bioconductordevtools::install_github()install an R package from Github.
.libPaths() reports the directory where your installed R packages are located.
Commenting
You can annotate your code by starting annotations with #. Comments to the right of # will be ignored by R.
Use # ---- to create navigable code sections.
For report generation, use R Markdown or Quarto.
Assignment operators
Anything that you want assigned to memory must be assigned to an R object.
<- the primary assignment operator, assigning values on the right to objects on the left.
= can also be used to assign values to objects, but is usually reserved for other purposes (e.g., function arguments)
Use ls() to list objects created in R. rm() can be used to remove an object from memory.
For R objects names,
avoid spaces or special characters, excluding "_" and ".".
do not begin with numbers or underscores.
* do not use names with special meanings (?Reserved).
Object data types
The base data type (e.g., numeric, character, logical, etc.) and the class (dataframe, matrix, etc.) will be important for what you can do with an object. Learn more about an object with the following:
class()returns the class of an object or base data typestr()returns the structure of an object.
dplyr::glimpse()
Similar to str() but with much more succinct output.
Coercion is when converting from one type to another, which may throw various warning messages. Always make sure output matches expectations.
Importing and exporting data
Use the read functions to import data (e.g., read.csv, read.delim, etc.). Use write functions to export data (e.g., write.table).
There are specific functions for unique data. For example, we will learn how to specifically import scRNA-seq data using Seurat.
Using functions
R functions perform specific tasks. R has a ton of built-in functions and functions available through additional packages. You can also create your own functions.
The general syntax for a function is the name followed by parentheses, function_name() (e.g., round()).
To create a function:
function_name <- function(arg_1, arg_2, ...) {
Function body
}
Vectors
A vector is a collection of values that are all of the same type (numbers, characters, etc.) --- datacarpentry.org
c()- used to combine elements of a vector
When you combine elements of different types in the same vector, they are forced into the same type via "coercion" (logical < numeric < character).
* length() - returns the number of elements in a vector
Use brackets to extract elements of a vector:
a <- 1:10
a[2]
Lists
Unlike vectors, lists can hold values of different types.
list(1, "apple", 3)
Data frames
Data frames hold tabular data comprised of rows and columns; they can be created using data.frame().
To understand more about the structure of an object and data frame, consider the following functions:
str()displays the structure of an object, not just data framesdplyr::glimpse()similar tostrbut applies to data frames and produces cleaner outputsummary()produces result summaries of the results of various model fitting functionsncol()returns number of columns in data framenrow()returns number of rows of data framedim()returns row and column numbersunique()returns a vector of with duplicates removed; also seedplyr::distinct()
We can subset data frames using bracket notation df[row,column]:
df<- data.frame(Counts=seq(1,5), animals=c("racoon","squirrel","bird","dog","cat"))
#to return just the animals column
df[,"animals"]
dplyr such as filter() for subsetting by row and select() for subsetting by column.
Plotting
There are 3 primary plotting systems with R: base R, ggplot2, and lattice. Data visualization functions from Seurat primarily use ggplot2 and can easily be customized by adding additional ggplot2 layers.
Check out the R Graph Gallery for data visualization examples and code.
Conditionals and Looping
See the attached resources on
Getting info on R Session
sessionInfo() Print version information about R, the OS and attached or loaded packages. This is useful for reporting methods for publication. Consider using the package renv to track and share exact versions of packages used for any given R script / project.
Resources for learning R
-
Other cheat sheets can be found here.
-
There is also a nice review here.
-
There are a ton of free tutorials. There are at least 230 Git repositories that focus on R training related to bioinformatics. Check out Glittr.
-
DataQuest and Coursera licenses are available to CCR and NIH researchers respectively. See here for more information.
BTEP lessons / courses
- R Introductory Series
- Data Wrangling with R
- Data Visualization with R
- Toward Reproducibility with R on Biowulf
- A Beginner's Guide to Troubleshooting R Code
Seurat Computational Requirements
Regarding computational requirements, rule of thumb is to keep it at 8 samples or less when running on a laptop. You may be able to stretch it to 10 samples, but any more than that strains the computer, especially when running batch correction or imaging. The imaging can be somewhat mitigated by printing the image to a file with the
pngor
Note
Using the package BPCells to manage SeuratObjects can make your analysis more computationally efficient. We will create a merged seurat object in this tutorial, which will be ~ 2.1 GB. Using BPCells, the same object is ~63.9 MB.
See this vignette for more information.
Working with R on Biowulf
Due to limits on computational resources, you may be interested in running your analysis on an HPC. Biowulf is the NIH high performance compute cluster. It has greater than 90k processors, and can easily perform large numbers of simultaneous jobs. Biowulf also includes greater than 600 pre-installed scientific software and databases.
You will need to know the basics of working on the command line to use Biowulf. Minimally, you should be able to navigate a directory tree using basic Unix commands. If the command line is foreign to you, check out this introduction.
Warning
The default version of R on Biowulf is currently 4.3.2 (module -r avail '^R$'), and the version of Seurat that complements this installation is v5.0.1.
Need a Biowulf Account?
If you do not already have a Biowulf account, you can obtain one by following the instructions here. NIH HPC accounts are available to all NIH employees and contractors listed in the NIH Enterprise Directory. Obtaining an account requires PI approval and a nominal fee of $35 per month. Accounts are renewed annually contingent upon PI approval.
When you apply for a Biowulf account you will be issued two primary storage spaces:
/home/$USER (16 GB)
/data/$USER (100 GB)
You may request more space in /data/$USER by filing an online storage request.
Accessing RStudio on Biowulf
If you plan to use RStudio to interactively analyze your data, RStudio Server, a browser-based interface very similar to the standard RStudio desktop environment, is the best option to avoid issues with lag.
Each RStudio session with RStudio Server will start fresh, so be sure to save your environment (save.image()) or save important objects (saveRDS()) before you exit the session.
Interactive Sessions
To work interactively with RStudio on Biowulf, you will need to request an interactive session using sinteractive.
When using R, you will need to include a few more options while obtaining your interactive session. For example, you will likely need space for temporary files (lscratch) and more memory (mem). You may also need additional CPUs (--cpus-per-task) if your workflow uses multi-threading. A sinteractive tunnel (--tunnel) will be required for RStudio Server on Biowulf.
Example interactive session request:
sinteractive --cpus-per-task=8 --gres=lscratch:50 --mem=50g --tunnel
NIH HPC Documentation
If you need help with Biowulf, the NIH HPC systems are well-documented at hpc.nih.gov. The User guides, Training documentation, and How To docs are also fantastic resources for getting help with most HPC tasks.
For all other questions, consider emailing the HPC team directly at staff@hpc.nih.gov.
Getting Started with Seurat
The Data
In this tutorial, we are using data from Nanduri et al. 2022, Epigenetic regulation of white adipose tissue plasticity and energy metabolism by nucleosome binding HMGN proteins, published in Nature Communications. The raw count matrices are available in GEO here, and the raw fastq files are available in the SRA.
An R project directory containing these data and associated files can be downloaded here.
This study examined "the role of HMGNs in white adipocyte browning by comparing wild-type (WT) mice and cells to genetically derived mice and cells lacking the two major members of the HMGN protein family, HMGN1 and HMGN2 (DKO mice)." HMGN proteins are known chromatin binding proteins that have been shown to play a role in cell identity stabilization via epigenetic regulation.
| Sample | Species | Condition | Time Point | Replicate | Status |
|---|---|---|---|---|---|
| W10 | Mouse | WT | 0 | 1 | Preadipocytes |
| W20 | Mouse | WT | 0 | 2 | Preadipocytes |
| D10 | Mouse | DKO | 0 | 1 | Preadipocytes |
| D20 | Mouse | DKO | 0 | 2 | Preadipocytes |
| W16 | Mouse | WT | 6 | 1 | Adipocytes |
| W26 | Mouse | WT | 6 | 2 | Adipocytes |
| D16 | Mouse | DKO | 6 | 1 | Adipocytes |
| D26 | Mouse | DKO | 6 | 2 | Adipocytes |
Experimental Design
The general experimental design was as follows:
WT vs DKO mice at two time points, 0 and 6 days. At day 0, cells were in a preadipocyte state, while at day 6 they had differentiated into adipocytes. Each time point had two replicates of each condition (WT vs DKO), for a total of 8 samples.
Installation
Seurat can be installed directly from CRAN.
install.packages("Seurat")
If you would like to install the development version or previous versions, see the installation instructions available here.
Load the package from your R library. We will also go ahead and load packages from the tidyverse.
library(Seurat)
library(tidyverse)
Importing Data
Now that we have loaded the package, we can import our data.
Generally, in R programming, functions that involve data import begin with "read / Read". Seurat includes a number of read functions for different data / platform types.
For 10X scRNA-Seq data, the following functions will be most relevant:
Read10X()- primary argument is a directory from CellRanger containing the matrix.mtx, genes.tsv (or features.tsv), and barcodes.tsv files.Read10X_h5()- reads the hdf5 file from 10X CellRanger (scATAC-Seq or scRNA-Seq).ReadMtx()- read from local or remote (arguments for the .mtx file, cells/barcodes file, and features/genes file). This option is not just for 10X data, but is useful for most platforms and pipelines.
The function you choose will depend on the format of your data.
Note
There are a number of other read and load functions available for different platforms and data types (e.g., Akoya CODEX, Nanostring SMI, SlideSeq, 10X Visium, 10X Xenium, Vizgen MERFISH). You can find relevant functions here.
Importing data in a .tsv or .csv
If you are trying to import publicly available data, and the available count data was saved as a .txt, .tsv, or .csv file, you can import these data using regular base R import functions (e.g., read.table(), read.csv()). The data does not need to be in a sparse format to create a Seurat object. However, if you would like to save memory upon import by loading as a sparse data matrix, see readSparseCounts() from the Bioconductor package scuttle.
Should I use the filtered for unfiltered data from Cell Ranger?
The raw / unfiltered matrix includes all valid cell barcodes from GEMs (Gel Bead-In EMulsions). This includes GEMs with free-floating mRNA from lysed or dead cells. Cell Ranger uses a barcode vs UMI count rank plot (knee plot) to eliminate such background noise.
While using the raw files will give you greater control over filtering, these files are also much larger. You can visualize the web summary output from Cell Ranger to see what was filtered. If you do not see a clear inflection point, you may want to use the raw files and apply other techniques for filtering.
Loading hdf5 files.
To load a single file, we use
W10 <- Read10X_h5("../data/W10_filtered_feature_bc_matrix.h5")
The only required argument is the path to the 10X h5 file.
To read multiple files, you can use a for loop or a function(s) from the apply family. For example,
#List files
files <- list.files(path="../data/",recursive=T,pattern="*.h5")
#Create a list of count matrices
h5_read <- lapply(paste0("../data/",
files), Read10X_h5)
#Automated way to assign names; modify for your purposes
#names(h5_read)<- sapply(files,
# function(x){str_split_1(x,"_")[1]},
# USE.NAMES = FALSE)
#Assign names manually
names(h5_read)<-c("D10","D16","D20","D26","W10","W16","W20","W26")
Let's take a look at the W10.
W10[1:5,1:5]
5 x 5 sparse Matrix of class "dgCMatrix"
AAACCCACAGTGACCC-1 AAACCCACATGGCTGC-1 AAACCCAGTGATATAG-1
Xkr4 . . .
Gm1992 . . .
Gm19938 . . 1
Gm37381 . . .
Rp1 . . .
AAACCCAGTGGAAGTC-1 AAACCCATCACGTCCT-1
Xkr4 . .
Gm1992 . .
Gm19938 . .
Gm37381 . .
Rp1 . .
The data is stored as a sparse matrix to save CPU, time, and memory. Each . is actually a 0.
Creating a Seurat Object
Once we have read in the matrices, the next step is to create a Seurat object. The Seurat object will be used to store the raw count matrices, sample information, and processed data (normalized counts, plots, etc.).
CreateSeuratObject() is used to create the object. This requires the matrix of counts for the first argument (counts=W10). We can also include a project name (e.g., the sample name). Other useful arguments include min.cells and min.features, which allow some initial filtering. For example, min.cells = 3 will filter genes / features that are not present across a minimum of 3 cells, while min.feature=200 will filter cells that do not contain a minimum of 200 genes / features.
# Single sample
W10 <- CreateSeuratObject(counts=W10, project="W10", min.cells = 3,
min.features = 200)
# View W10
W10
An object of class Seurat
19059 features across 7382 samples within 1 assay
Active assay: RNA (19059 features, 0 variable features)
1 layer present: counts
For all samples, we can use mapply to loop through the h5_read list and the list names (names(h5_read)).
# All samples
adp <- mapply(CreateSeuratObject,counts=h5_read,
project=names(h5_read),
MoreArgs = list(min.cells = 3, min.features = 200))
# View adp
adp
$D10
An object of class Seurat
19084 features across 7623 samples within 1 assay
Active assay: RNA (19084 features, 0 variable features)
1 layer present: counts
$D16
An object of class Seurat
16943 features across 5990 samples within 1 assay
Active assay: RNA (16943 features, 0 variable features)
1 layer present: counts
$D20
An object of class Seurat
18242 features across 5093 samples within 1 assay
Active assay: RNA (18242 features, 0 variable features)
1 layer present: counts
$D26
An object of class Seurat
17751 features across 7436 samples within 1 assay
Active assay: RNA (17751 features, 0 variable features)
1 layer present: counts
$W10
An object of class Seurat
19059 features across 7382 samples within 1 assay
Active assay: RNA (19059 features, 0 variable features)
1 layer present: counts
$W16
An object of class Seurat
17024 features across 5465 samples within 1 assay
Active assay: RNA (17024 features, 0 variable features)
1 layer present: counts
$W20
An object of class Seurat
18354 features across 5427 samples within 1 assay
Active assay: RNA (18354 features, 0 variable features)
1 layer present: counts
$W26
An object of class Seurat
17373 features across 6013 samples within 1 assay
Active assay: RNA (17373 features, 0 variable features)
1 layer present: counts
#remove the original sparse matrices
rm(h5_read)
We can merge our seurat objects, which are now in a list in order to view the QC metrics and compare across samples. If samples are fairly different, QC thresholds should be applied on a per sample basis.
adp <- merge(adp[[1]], y = adp[2:length(adp)],
add.cell.ids = names(adp),project="Adipose")
The cell IDs can overlap between samples, so here we can apend the sample ID as a prefix to each cell ID using add.cell.ids. If this isn't done "and any cell names are duplicated, cell names will be appended with _X, where X is the numeric index of the object in c(x, y)" (See merge.Seurat()).
Warning
We could have created a merged object using h5_read, as a list can be provided for the argument counts:
adp2<-CreateSeuratObject(counts=h5_read, min.cells = 3,
min.features = 200)
However, this seems to take longer and does not give us control over the cell IDs. The documentation for using a list with CreateSeuratObject() is currently lacking. It is unclear how to use the metadata option if cell ids are not unique across samples.
Use a for loop to load the data and create an object for each file
If you want to apply QC metrics independently for each sample, you can use this for loop to create an individual object for each sample.
# Create a Seurat object for each sample
for (file in files){
seurat_data <- Read10X_h5(paste0("../data/",
file))
seurat_obj <- CreateSeuratObject(counts = seurat_data,
min.features = 200,
min.cells = 3,
project = file)
assign(file, seurat_obj)
}
See here for an explanation of the code.
Inspect and work with a Seurat object
The Seurat Object is a data container for single cell RNA-Seq and related data. It is an S4 object, which is a type of data structure that stores complex information (e.g., scRNA-Seq count matrix, associated sample information, and data /results generated from downstream analyses) in one or more slots. It is extensible and can interact with many different methods, including those from other developers.
You can see the primary slots using glimpse().
glimpse(W10)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
.. ..$ RNA:Formal class 'Assay5' [package "SeuratObject"] with 8 slots
..@ meta.data :'data.frame': 7382 obs. of 3 variables:
.. ..$ orig.ident : Factor w/ 1 level "W10": 1 1 1 1 1 1 1 1 1 1 ...
.. ..$ nCount_RNA : num [1:7382] 23082 35379 17308 27507 2858 ...
.. ..$ nFeature_RNA: int [1:7382] 4760 6122 4697 5526 995 4261 4089 1744 6004 5872 ...
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 1 level "W10": 1 1 1 1 1 1 1 1 1 1 ...
.. ..- attr(*, "names")= chr [1:7382] "AAACCCACAGTGACCC-1" "AAACCCACATGGCTGC-1" "AAACCCAGTGATATAG-1" "AAACCCAGTGGAAGTC-1" ...
..@ graphs : list()
..@ neighbors : list()
..@ reductions : list()
..@ images : list()
..@ project.name: chr "W10"
..@ misc : list()
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
.. ..$ : int [1:3] 5 0 1
..@ commands : list()
..@ tools : list()
W10 has 13 slots:
assays, meta.data, active.assay, active.ident, graphs, neighbors, reductions, images, misc, version, commands, and tools. See a description of the slots here. For guidance on accessing data stored in the SeuratObject, see this vignette.
Unlike previous iterations of Seurat, Seurat v5 contains assays in layers to accommodate different assays and data types. "For example, these layers can store: raw counts (layer='counts'), normalized data (layer='data'), or z-scored/variance-stabilized data (layer='scale.data')."
To return the names of the layers:
Layers(W10)
[1] "counts"
To access a specific count layer use:
W10[["RNA"]]$counts |> head()
# or
W10@assays$RNA$counts |> head()
# or
LayerData(W10, assay="RNA", layer='counts') |> head()
Because we have just created this object, most of the slots are empty. We will save results back to the object and fill the slots as we work.
You can always check the version of Seurat that was used to build the object with
W10@version
[1] '5.0.1'
And check commands and parameters used to generate results with:
W10@commands
list()
We haven't worked on the object, so there is nothing here. Compare with pbmc_small a built in data set in the SeuratObject package.
head(pbmc_small@commands, 1)
$NormalizeData.RNA
Command: NormalizeData(object = pbmc_small)
Time: 2018-08-27 16:32:17.136699
assay : RNA
normalization.method : LogNormalize
scale.factor : 10000
verbose : TRUE
This information is useful for documenting your analysis.
Examine metadata
The metadata in the Seurat object is located in adp@metadata and contains the information associated with each cell.
We can access the metadata using:
head(adp@meta.data) #using head to return only the first 6 rows
orig.ident nCount_RNA nFeature_RNA
D10_AAACCCAAGATGCTTC-1 D10 11188 3383
D10_AAACCCAAGCTCTTCC-1 D10 32832 5823
D10_AAACCCAAGTCATCGT-1 D10 28515 5002
D10_AAACCCAGTAGCGTCC-1 D10 18954 4305
D10_AAACCCAGTGCACGCT-1 D10 27181 4586
D10_AAACCCAGTGTAAATG-1 D10 3090 1030
or
head(adp[[]])
orig.ident nCount_RNA nFeature_RNA
D10_AAACCCAAGATGCTTC-1 D10 11188 3383
D10_AAACCCAAGCTCTTCC-1 D10 32832 5823
D10_AAACCCAAGTCATCGT-1 D10 28515 5002
D10_AAACCCAGTAGCGTCC-1 D10 18954 4305
D10_AAACCCAGTGCACGCT-1 D10 27181 4586
D10_AAACCCAGTGTAAATG-1 D10 3090 1030
Accessing a column or columns:
#Access a single column.
head(adp$orig.ident)
D10_AAACCCAAGATGCTTC-1 D10_AAACCCAAGCTCTTCC-1 D10_AAACCCAAGTCATCGT-1
"D10" "D10" "D10"
D10_AAACCCAGTAGCGTCC-1 D10_AAACCCAGTGCACGCT-1 D10_AAACCCAGTGTAAATG-1
"D10" "D10" "D10"
head(adp[["orig.ident"]])
orig.ident
D10_AAACCCAAGATGCTTC-1 D10
D10_AAACCCAAGCTCTTCC-1 D10
D10_AAACCCAAGTCATCGT-1 D10
D10_AAACCCAGTAGCGTCC-1 D10
D10_AAACCCAGTGCACGCT-1 D10
D10_AAACCCAGTGTAAATG-1 D10
#Access multiple columns, rows.
head(adp[[c("orig.ident", "nCount_RNA")]])[1:3,]
orig.ident nCount_RNA
D10_AAACCCAAGATGCTTC-1 D10 11188
D10_AAACCCAAGCTCTTCC-1 D10 32832
D10_AAACCCAAGTCATCGT-1 D10 28515
As we can see, there are different ways to access and work with the metadata. In addition, we can see that we begin with built-in information such as the orig.ident, nCount_RNA, and nFeature_RNA.
orig.ident - defaults to the project labels
nCount_RNA - number of UMIs per cell
nFeature_RNA - number of genes / features per cell
This information, along with other metrics, will be used for quality filtering.
Seurat Idents?
Idents are the metadata used by Seurat to classify each of the datapoints. Each datapoint can have multiple metadata identities (e.g., sample, condition, cluster, etc.). The active.ident is the default Ident used at even given time by a particular Seurat function. Often it can be adjusted using function parameters; however, at other times, you will need to actively change the active.ident to another metadata column using Idents(). See ?Idents() and this vignette for examples.
Subsetting the object
We can subset by features or cells by adp[1:10,] or adp[,1:10] respectively. To subset using metadata, use subset().
For example, if we want to subset to the sample W10, we can use.
subset(x = adp, idents = "W10")
An object of class Seurat
19059 features across 7382 samples within 1 assay
Active assay: RNA (19059 features, 0 variable features)
1 layer present: counts.W10
We will look at subsetting more when filtering cells based on quality.
Add to metadata.
We can add information to our metadata by accessing and assigning to metadata columns or using ?AddMetaData().
Add condition to metadata (either wildtype of double knockout).
adp$condition <- ifelse(str_detect(adp@meta.data$orig.ident, "^W"),
"WT","DKO")
Add time information to the metadata.
adp$time_point <- ifelse(str_detect(adp@meta.data$orig.ident, "0"),
"Day 0","Day 6")
Add Condition + Time to the metadata.
adp$cond_tp <- paste(adp$condition, adp$time_point)
Other Useful information from the Seurat object
The Seurat v5 object doesn’t require all assays have the same cells. In this case, Cells() can be used to return the cell names of the default assay while colnames() can be used to return the cell names of the entire object.
head(Cells(adp,layer="counts.W10"))
[1] "W10_AAACCCACAGTGACCC-1" "W10_AAACCCACATGGCTGC-1" "W10_AAACCCAGTGATATAG-1"
[4] "W10_AAACCCAGTGGAAGTC-1" "W10_AAACCCATCACGTCCT-1" "W10_AAACGAACAGGCCTGT-1"
head(colnames(adp))
[1] "D10_AAACCCAAGATGCTTC-1" "D10_AAACCCAAGCTCTTCC-1" "D10_AAACCCAAGTCATCGT-1"
[4] "D10_AAACCCAGTAGCGTCC-1" "D10_AAACCCAGTGCACGCT-1" "D10_AAACCCAGTGTAAATG-1"
To return feature/gene names:
head(Features(adp))
[1] "Xkr4" "Gm19938" "Sox17" "Mrpl15" "Lypla1" "Tcea1"
head(rownames(adp))
[1] "Xkr4" "Gm19938" "Sox17" "Mrpl15" "Lypla1" "Tcea1"
For multimodal data, list the assays using
Assays(adp)
[1] "RNA"
To return the number of cells or features use:
#return number of cells across all layers
num_cells <- ncol(adp)
#return number of features across all layers
num_features <- nrow(adp)
#to return row by column information
dim(adp)
[1] 20654 50429
Note
ncol(adp) will return the number of cells across all layers. If you want a specific layer, use ncol(adp[["RNA"]]$counts.W10). This information is also quickly accessible via the Global Environment when using RStudio or by using str().
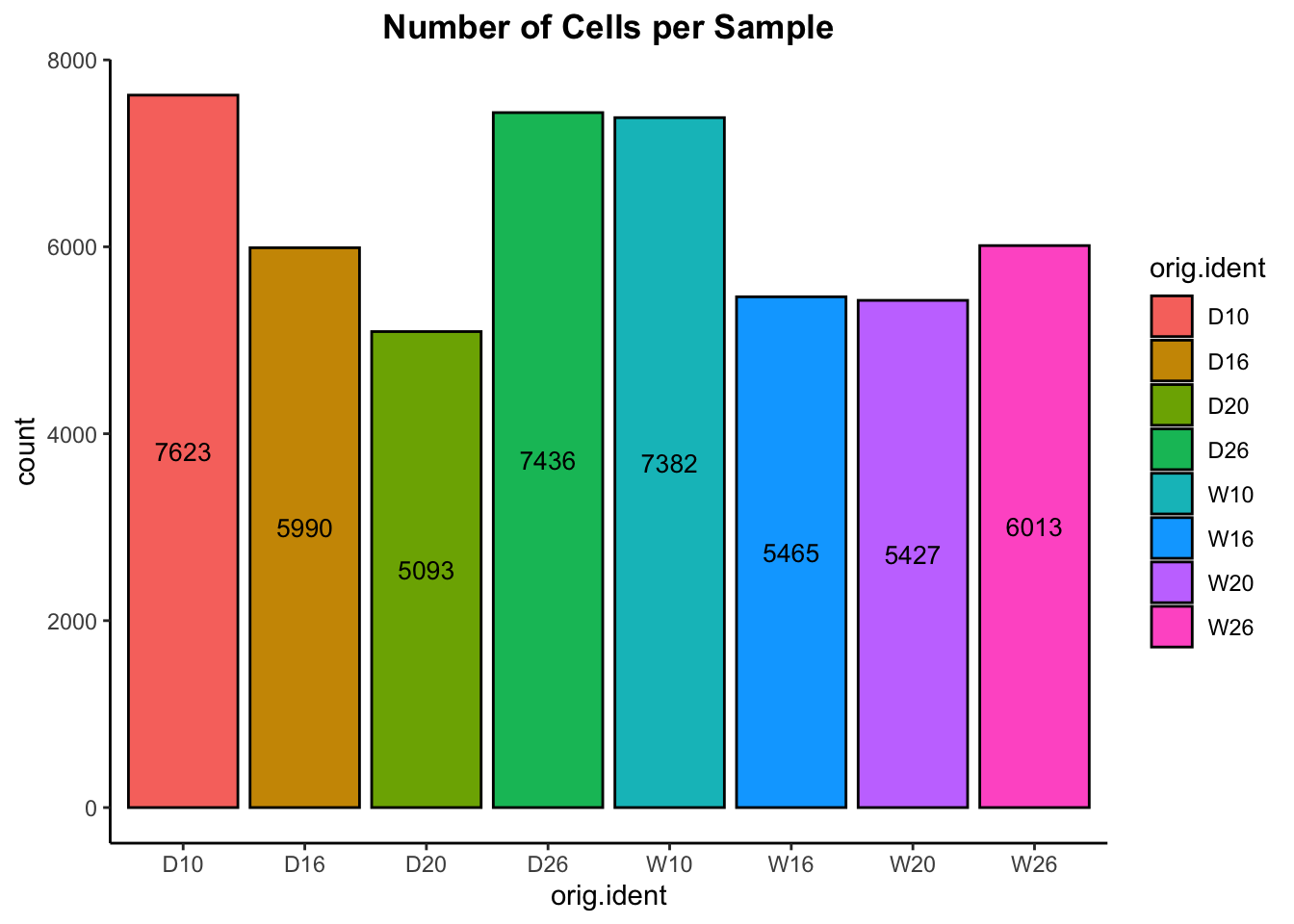
We can also quickly plot the number of cells from the metadata:
# Visualize the number of cell counts per sample
adp@meta.data %>%
ggplot(aes(x=orig.ident, fill=orig.ident)) +
geom_bar(color="black") +
stat_count(geom = "text", colour = "black", size = 3.5,
aes(label = ..count..),
position=position_stack(vjust=0.5))+
theme_classic() +
theme(plot.title = element_text(hjust=0.5, face="bold")) +
ggtitle("Number of Cells per Sample")
Warning: The dot-dot notation (`..count..`) was deprecated in ggplot2 3.4.0.
ℹ Please use `after_stat(count)` instead.
Save the object
At this point, our Seurat object is fairly large (~2.1 GB). It is wise to save this for downstream applications using saveRDS().
saveRDS(adp,"../outputs/merged_Seurat_adp.rds")
References
The following resources were instrumental in designing this lesson: