nf-core is a community effort to generate a curated set of standardized, best-practice, reproducible, documented, NGS analysis pipelines. All these workflows are built using the versatile workflow manager, Nextflow, and have been released under the MIT license. This process has resulted in workflows that all have the following characteristics:
- Integrated software dependency management (Docker, Singularity [now called Apptainer], Conda).
- Portability to run your pipelines anywhere: laptop, cluster, or cloud.
- Reproducibility of analyses independent of time and computing platform.
- There is only a single pipeline per data/analysis type.

At the time of this writing there are 53 released pipelines, with another 23 under development. This large collection of pipelines, ranging from RNA-Seq to spatial transcriptomics, address most standard NGS technologies.
Even though these pipelines have been built to be user-friendly and combine many different steps into a single command, they are nonetheless complicated pieces of software which, for effective use, typically require:
- Access to and familiarity with the Unix computer architecture and file systems
- An understanding of the concepts of containers (Docker/Singularity/Conda)
- Familiarity with the steps involved in the analysis of a given experimental protocol (e.g. RNASEQ).
- Access to the appropriate genome sequence data (sequences and annotations) for the desired genome version. Natively the nf-core pipelines make use of data residing in the “Cloud” within the iGenomes project, which unfortunately, has not been kept current (especially with respect to annotations).
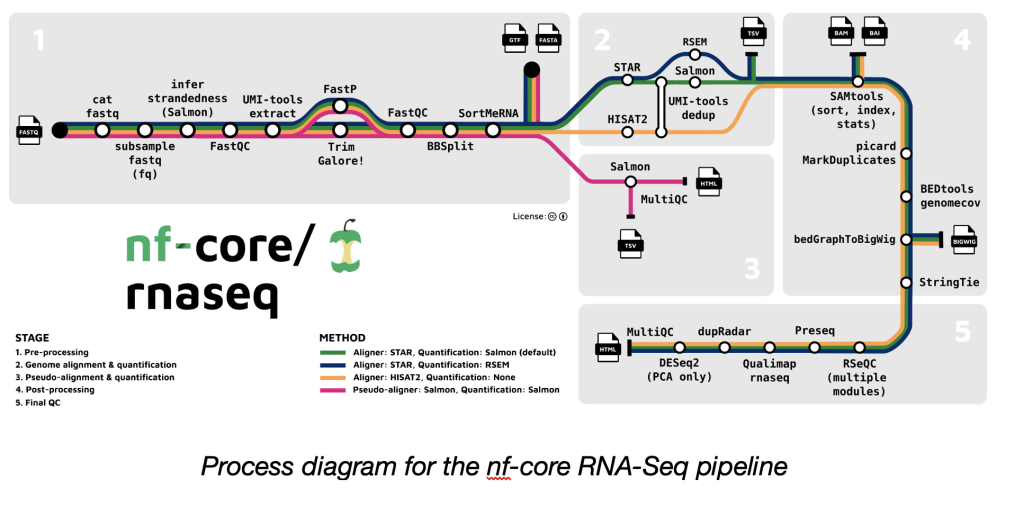
CCR scientists can access the nf-core pipelines in two distinct environments: the NIH HPC system Biowulf and the Genome Analysis Unit (GAU) sponsored DNAnexus Cloud Platform.
The NIH HPC implementation makes use of Biowulf’s slurm task management software for job scheduling and the Singularity container software to stage the necessary software tools. This implementation allows access to the entire suite of pipelines. While the HPC documentation on running nf-core pipelines is minimal, the pages devoted to Nextflow(https://hpc.nih.gov/apps/nextflow.html) should serve as a starting point.

Those who are not comfortable working at the command line or do not have experience working with unfamiliar software should consider the DNAnexus option described below.
nf-core on DNAnexus
As mentioned above, nf-core pipelines in their native format, present some challenges for those who are not “professional computational biologists“. To facilitate the use of these pipelines by the less skilled users, the Genome Analysis Unit (GAU) has implemented the most common nf-core pipelines within the DNAnexus Cloud environment. This implementation offers several advantages.
- A simple to use Grapical User Interface (GUI)… point and click.
- Ready access to a Cloud network of computation resources for faster execution via distributed network of processing power.
- Integrated documentation offering a guided approach to each analysis.
- Pre-indexed versions of the most common genomes (human, mouse).
Additionally, GAU has taken the approach of presenting each of these workflows in two formats:
- The simplified interface – Offering a pared down set of user choices, a guided selection of available genomes (organism and version), simplified input and enhanced output. This enhanced output include highlighting the most important output and providing a IGV session file to allow direct viewing of generated bam and bigwig files (without the need to download the files directly).
- The full interface – for sophisticated users who want to access the full set of choices available within the workflow. This version presents the native workflow with all its options, available via either the DNAnexus GUI or CLI interfaces.
In part two of this article we provide more details about:
- How to get an account and access these pipeline on the DNAnexus Cloud Platform.
- A more detailed description of the pipelines available, and how to use them as well as links to on-line documentation.
- Dates for upcoming training sessions on the use of nf-core pipelines on DNAnexus.
STAY TUNED!
— Peter FitzGerald & Carl McIntosh (Genome Analysis Unit)
This topic spotlight was authored on October 3, 2023 and reflects the information available at that time. As content may evolve, please contact us at ncibtep@nih.gov to obtain the most current information.