Interactive and Batch Jobs on Biowulf
Learning Objectives
After this class, the participant should have obtained the foundations for performing compute intensive tasks on Biowulf. Specifically, the participant should be able to
- Describe Biowulf and high performance computing (HPC) systems
- Work interactively on Biowulf compute nodes
- Submit shell and swarm scripts to the Biowulf batch system
Background on Biowulf
What is Biowulf
Biowulf is the Unix/Linux-based high performance computing (HPC) system at NIH. With over 90,000 processor cores and 30 petabytes of storage space, it is much more powerful than a personal computer. As depicted in Figure 1, a high performance computing system is composed of a cluster of individual computers. Each computer has it's own processors, data storage, and random access memory (RAM). The individual computers are referred to as nodes. It is beneficial to know the terminologies in Figure 1 as these are encountered when working on Biowulf or other high performance computing systems.
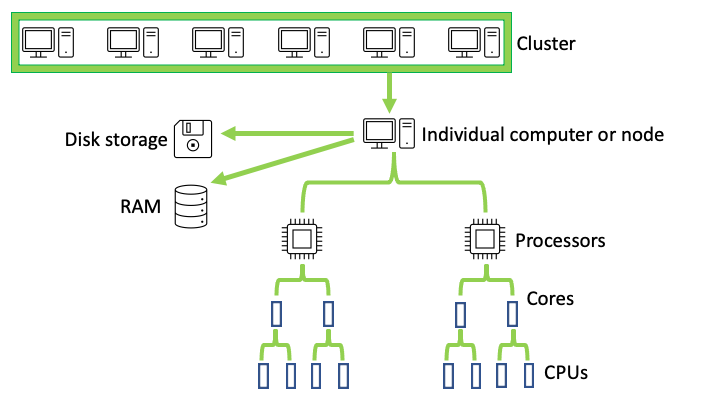
Figure 1: Example of high performance computing structure.
How does Biowulf manage and allocate compute resources?
Biowulf and other HPC systems use SLURM (Simple Linux Utility for Resource Management) to manage and allocate computation resources.
Note
"Slurm allocates on the basis of cores." -- Biowulf swarm tutorial
Why use Biowulf?
- Compute power
- Many bioinformatics as well scientific software
- System and software maintenance performed by Biowulf staff
- Option for users to install their own software
- Transfer large datasets
Interactive sessions
Upon connecting to Biowulf, the user lands in the login node.
Warning
Do not use the login node to perform any computation intensive tasks on Biowulf. Instead, we should either submit a job (if staying in the log in node) with sufficient resources requested or request an interactive session if we are going to be doing some testing and development.
The log in node is meant for the following (Source: Biowulf interactive jobs)
- Submitting jobs (main purpose)
- Editing/compiling code
- File management
- File transfer
- Brief testing of code or debugging (about 20 minutes or less)
Interactive sessions allows users to work on Biowulf compute nodes and are suitable for
- Testing/debugging cpu-intensive code
- Pre/post-processing of data
- Use of graphical application
Note
Always work in the data directory!
Use pwd to check if the present working directory is /data/username, where username is user's Biowulf account ID. If not, then change into it by using cd /data/username.
To request an interactive session with default resource allocations use
sinteractive
Biowulf will search for available resources and allocate. The number 66874095 below is ID assigned specifically to this interactive job. If the user terminates the interactive session and requests a new one, a different job ID will be assigned to that interactive session.
salloc: Pending job allocation 66874095
salloc: job 66874095 queued and waiting for resources
salloc: job 66874095 has been allocated resources
salloc: Granted job allocation 66874095
salloc: Waiting for resource configuration
salloc: Nodes cn4271 are ready for job
To find how much resources have been allocated, use jobhist followed by the job id.
jobhist 66874095
Figure 2 shows an example of the output from jobhist command.
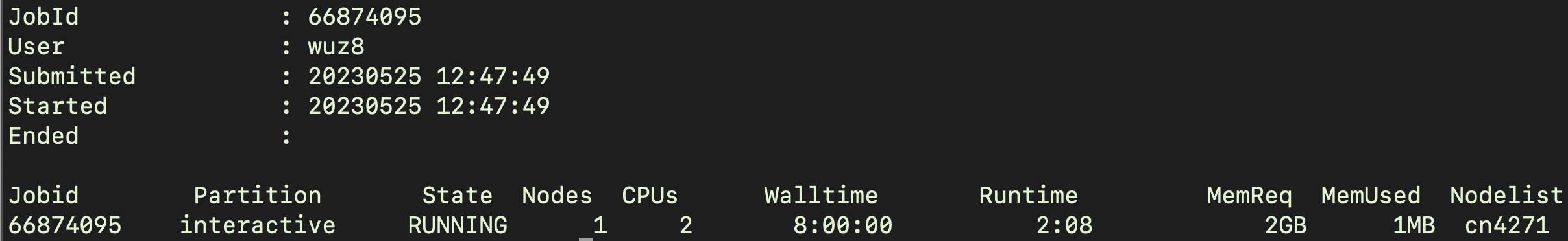
Figure 2: The default sinteractive allocation is 1 core (2 CPUs) and 0.768 GB/CPU (1.536 GB but rounded to 2 GB in the terminal) of memory and a walltime of 8 hours.
When done with the interactive session
exit
When done with Biowulf
exit
Options for sinteractive can be found at https://hpc.nih.gov/docs/userguide.html#int. The options include
--mem: to specify the amount of memory--gres: to ask for generic resources like temporary storage space--constraint: to specify the partition to use
"NCI-CCR has funded 153 nodes (4548 physical cores, 9096 cpus with hyperthreading) in the Biowulf cluster, and CCR users have priority access to these nodes." -- Biowulf NCI CCR partition
To use the ccr partition with an interactive session
sinteractive --constraint=ccr
Note
Each node in Biowulf has space that could be used to store temporary data (lscratch). These can be used for applications that write temporary files. To request lscratch space, include the --gres option in sinteractive.
Requesting an interactive session with
- 15 gb of temporary storage space
- 6 gb of memory
sinteractive --gres=lscratch:15 --mem=6gb
Note
Remember to use module load to activate a software before using it.
Download sequence data from the Sequence Read Archive using fastq-dump from the sratoolkit.
module load sratoolkit
The options and argument for the fastq-dump command below are
-X: input the number sequences to be downloaded from a FASTQ file, this example downloads the 200000 sequences.--skip-technical: do not download any technical sequences.--split-files: this option is for paired end sequencing and will download the forward and reverse reads into separate files.- Argument: the SRA sequence accession (ie. SRR23341296, which is derived from a RNA sequencing study)
fastq-dump -X 200000 --skip-technical --split-files SRR23341296
Load seqkit to get some statistics about the downloaded sequences using its stats module.
module load seqkit
seqkit stats SRR23341296*.fastq
file format type num_seqs sum_len min_len avg_len max_len
SRR23341296_1.fastq FASTQ DNA 200,000 30,000,000 150 150 150
SRR23341296_2.fastq FASTQ DNA 200,000 30,000,000 150 150 150
Use HISAT2, a splice aware aligner to map SRR23341296_1.fastq and SRR23341296_2.fastq to human genome. SRR23341296_1.fastq and SRR23341296_2.fastq are derived from RNA sequencing, thus require a splice aware aligner.
module load hisat
The options and arguments for the hisat2 command below are
-x: to specify the path of the reference genome (ie. $HISAT_INDEXES/grch38/genome)-1: "Comma-separated list of files containing mate 1s (filename usually includes _1), e.g. -1 flyA_1.fq,flyB_1.fq. Sequences specified with this option must correspond file-for-file and read-for-read with those specified in<m2>. Reads may be a mix of different lengths. If - is specified, hisat2 will read the mate 1s from the “standard in” or “stdin” filehandle." -- HISATS2 manual-2: "Comma-separated list of files containing mate 2s (filename usually includes _2), e.g. -2 flyA_2.fq,flyB_2.fq. Sequences specified with this option must correspond file-for-file and read-for-read with those specified in<m1>. Reads may be a mix of different lengths. If - is specified, hisat2 will read the mate 2s from the “standard in” or “stdin” filehandle." -- HISATS2 manual-S: specify name of the alignment output- Arguments:
- Files to be aligned (SRR23341296_1.fastq and SRR23341296_2.fastq)
- Name of the alignment output (SRR23341296.sam)
hisat2 -x $HISAT_INDEXES/grch38/genome -1 SRR23341296_1.fastq -2 SRR23341296_2.fastq -S SRR23341296.sam
200000 reads; of these:
200000 (100.00%) were paired; of these:
13538 (6.77%) aligned concordantly 0 times
178979 (89.49%) aligned concordantly exactly 1 time
7483 (3.74%) aligned concordantly >1 times
----
13538 pairs aligned concordantly 0 times; of these:
763 (5.64%) aligned discordantly 1 time
----
12775 pairs aligned 0 times concordantly or discordantly; of these:
25550 mates make up the pairs; of these:
17752 (69.48%) aligned 0 times
6942 (27.17%) aligned exactly 1 time
856 (3.35%) aligned >1 times
95.56% overall alignment rate
Find out job information such as resources allocated and used in the Biowulf user dashboard. See Figure 3 for an example of job information found in the user dashboard.
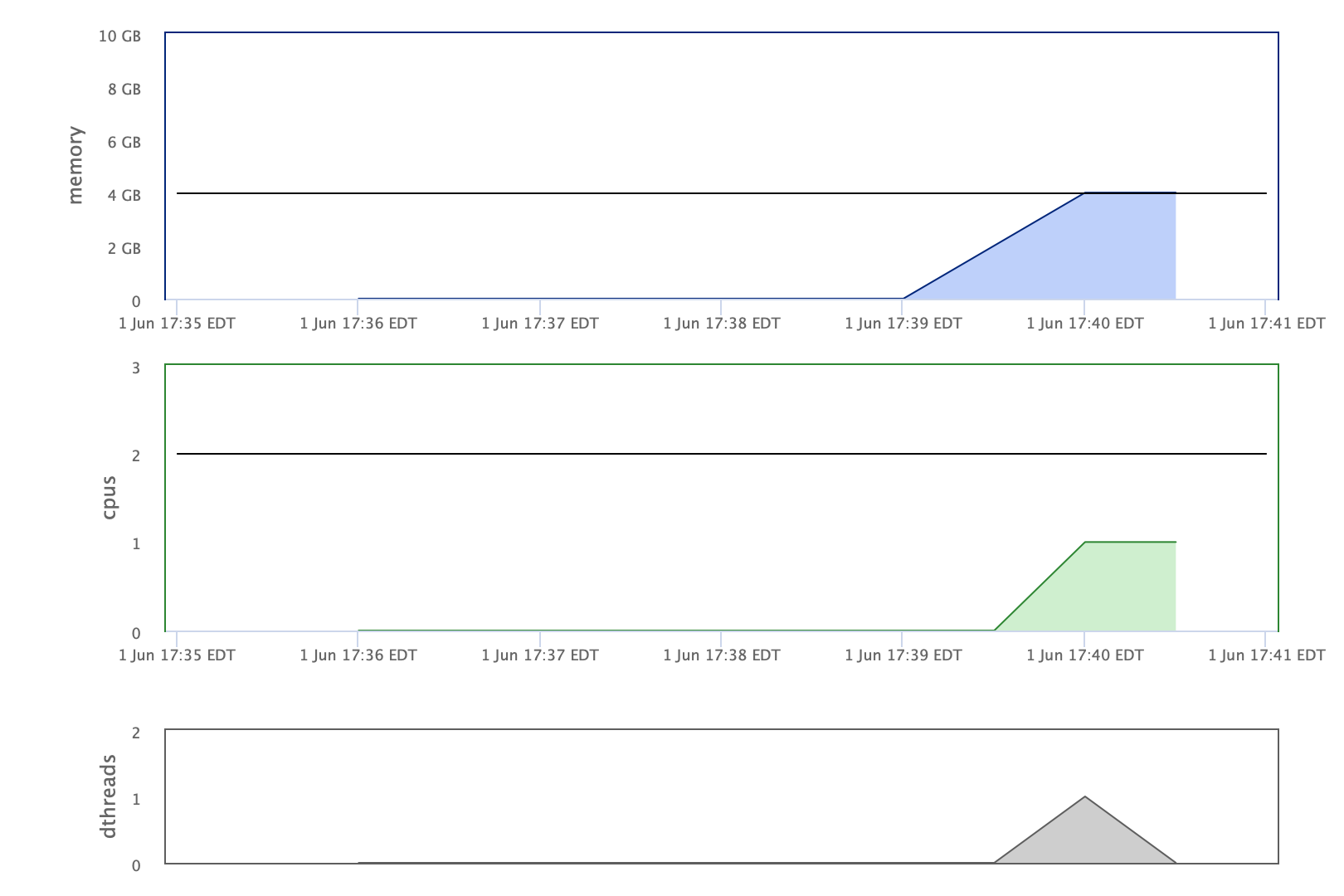
Figure 3: Users can find information regarding resources allocated and used in a job that was submitted to the Biowulf batch system via the user dashboard. The important statistics include memory allocated and used (top panel) as well as cpus allocated and used (middle panel). This example was derived from a hisat2 RNA sequencing alignment running on an interactive session with 4gb of memory requested (sinteractive --gres =lscratch:15 --mem=4gb). The memory usage was reaching the allocation limit and this alignment did not run successfully.
Submitting jobs to the Biowulf batch system
Users can test and run commands one by one on a interactive session. However, this is not practical and makes it hard for users to document analyses steps.
Shell script
A script allows users to string together analysis steps and keep them in one document. It can be reused with or without modification to analyze similar datasets.
The script (SRR23341296_statistics.sh) below will use seqkit stat to generate statistics for SRR23341296_1.fastq and SRR23341296_2.fastq. Important features of the shell scripts are listed below.
- They have extension
.sh - They start with #!/bin/bash
- "#!" is known as the sha-bang
- following #! is the path to the command interpreter (ie. /bin/bash)
- Lines that begin with #SBATCH are not run as a part of the script but these are called SLURM directives, which provides critical information regarding the job submission, including
- Name of the job (
--job-name) - What types of job status notifications to send (
--mail-type) - Where to send job status notification (
--mail-user) - Memory to allocate (
--mem) - Time to allocate (
--time) - Name of the job output log (
--ouput)
- Name of the job (
- To add a comment, start a line with #. Comments are not run as a part of the script
Other options for SLURM directives can be found on the Biowulf website
#!/bin/bash
#SBATCH --job-name=SRR23341296_statistics
#SBATCH --mail-type=ALL
#SBATCH --mail-user=wuz8@nih.gov
#SBATCH --mem=2gb
#SBATCH --time=00:05:00
#SBATCH --output=SRR23341296_statistics_log
# Step 1: load modules
module load seqkit
# Step 2: get statistics for SRR23341296_1.fastq and SRR23341296_2.fastq
## A "for" loop is used to loop through fastq files with the SRR23341296 pattern
## The statistics for each fastq file will be written to SRR23341296_statistics.txt (note >> writes and appends to a file)
for file in SRR23341296*.fastq;
do
seqkit stat $file >> SRR23341296_statistics.txt;
done
To submit the script
sbatch SRR23341296_statistics.sh
The job ID assigned is 2763496
To check on job status use the jobhist command, followed by the job ID. Figure 4 shows the results of the jobhist output.
jobhist 2763496
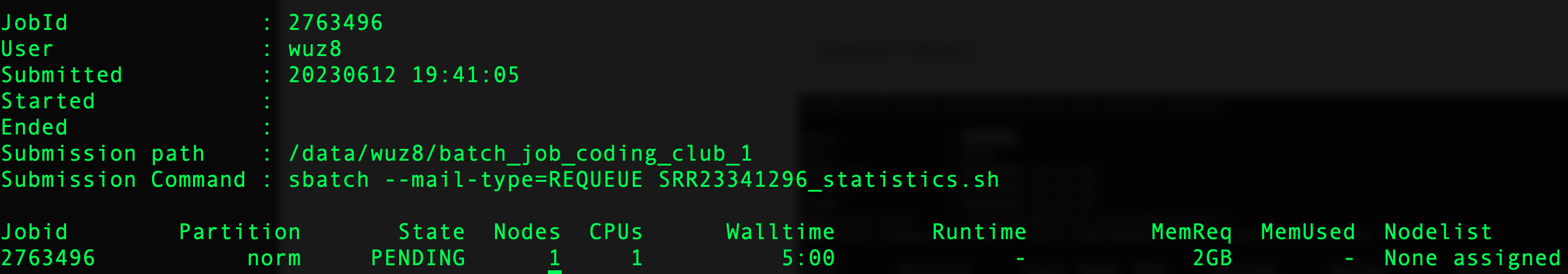
Figure 4: The jobhist commands retrieve status as well as resource allocated for a job. Note the status of job is PENDING as indicated by the "State" column. Biowulf will update the job status to running or complete accordingly.
To cancel a job
scancel job_id
cat SRR23341296_statistics.txt
file format type num_seqs sum_len min_len avg_len max_len
SRR23341296_1.fastq FASTQ DNA 200,000 30,000,000 150 150 150
file format type num_seqs sum_len min_len avg_len max_len
SRR23341296_2.fastq FASTQ DNA 200,000 30,000,000 150 150 150
The shell script shown below (hbr_uhr.sh) analyzes RNA sequencing data derived from the Human Brain Reference (hbr) and Universal Human Reference (uhr) study. It aligns sequences to genome, calculates gene expression counts from the alignment results, and identifies differentially expressed genes and is an example of how a script can be used for multi-step analyses. In the script below, two additional sbatch directives were included.
--cpus-per-task: request a specified number of cpus for the job (6 in this case)gres=lscratch:5: to request 5 gb of temporary storage space. The path to the temporary storage space was exported as the variable TMPDIR usingexport TMPDIR=/lscratch/$SLURM_JOB_ID, where $SLURM_JOB_ID is the variable that is storing the ID of a job.
#!/bin/bash
#SBATCH --job-name=hbr_uhr
#SBATCH --mail-type=ALL
#SBATCH --mail-user=wuz8@nih.gov
#SBATCH --mem=4gb
#SBATCH --cpus-per-task=6
#SBATCH --time=00:10:00
#SBATCH --output=hbr_uhr_log
#SBATCH --gres=lscratch:5
export TMPDIR=/lscratch/$SLURM_JOB_ID
## This script will perform differential experssion analysis on the HBR/UHR dataset
## Step 1: load modules
module load hisat
module load samtools
module load subread
module load R
## Step 2: create folder for output
mkdir hbr_uhr_output
## Step 3: align sequencing reads to genome using hisat2
cat hbr_uhr/reads/ids.txt | parallel -j 6 "hisat2 -x hbr_uhr/refs/22 -1 hbr_uhr/reads/{}_R1.fq -2 hbr_uhr/reads/{}_R2.fq -S hbr_uhr_output/{}.sam"
## Step 4: convert alignment ouput sam file to machine readable bam file
cat hbr_uhr/reads/ids.txt | parallel -j 6 "samtools sort -o hbr_uhr_output/{}.bam hbr_uhr_output/{}.sam"
cat hbr_uhr/reads/ids.txt | parallel -j 6 "samtools index -b hbr_uhr_output/{}.bam hbr_uhr_output/{}.bam.bai"
## Step 5: obtain expression counts table from alignment results
featureCounts -p -a hbr_uhr/refs/22.gtf -g gene_name -o hbr_uhr_output/hbr_uhr_counts.txt hbr_uhr_output/HBR_1.bam hbr_uhr_output/HBR_2.bam hbr_uhr_output/HBR_3.bam hbr_uhr_output/UHR_1.bam hbr_uhr_output/UHR_2.bam hbr_uhr_output/UHR_3.bam
## Step 6: wrangle the expression counts results to pass onto differential expression analysis
sed '1d' hbr_uhr_output/hbr_uhr_counts.txt | cut -f1,7-12 | awk '{gsub(/hbr_uhr_output\//,""); print}' | tr '\t' ',' > hbr_uhr_output/counts.csv
## Step 7: perform differential expression analysis
cp hbr_uhr/design.csv ./hbr_uhr_output/design.csv
cd hbr_uhr_output
Rscript ../code/deseq2.r
To submit the script
sbatch hbr_uhr.sh
Swarm
Swarm is a way to submit multiple commands to the Biowulf batch system and each command will be run as an independent job, allowing for parallelization. Below is an example of swarm script (srp045416_download.swarm) that uses fastq-dump from the sratoolkit to download next generation sequencing data from the Sequence Read Archive (SRA).
- Swarm scripts have extension *.swarm
- Lines that start with #SWARM are not run as a part of the script but these are directives that tell the Biowulf batch system what resources (ie. memory, time, temporary storage, modules) are needed
#SWARM --job-name SRP045416
#SWARM --sbatch "--mail-type=ALL --mail-user=wuz8@nih.gov"
#SWARM --gres=lscratch:15
#SWARM --module sratoolkit
fastq-dump --split-files -X 10000 SRR1553606 -O SRP045416_fastq
fastq-dump --split-files -X 10000 SRR1553416 -O SRP045416_fastq
fastq-dump --split-files -X 10000 SRR1553417 -O SRP045416_fastq
fastq-dump --split-files -X 10000 SRR1553418 -O SRP045416_fastq
fastq-dump --split-files -X 10000 SRR1553419 -O SRP045416_fastq
Before submitting the swarm script, make a new directory called SRP045416_fastq to store the FASTQ files.
mkdir SRP045416_fastq
To submit a swarm script, use the swarm command where -f prompts for the name of the script.
swarm -f srp045416_download.swarm
The ID assigned to the swarm job is
2755884
To check on job status (see Figure 5 for output).
jobhist 2755884

Figure 5. jobhist output for the swarm script. Note that this screen capture was taken when the job was completed as indicated by the COMPLETED status in the "State" column. This swarm script created five sub-jobs labeled "2755884_01" thru "2755884_4" (see Jobid column). Each of the sub-jobs was assigned 2 gb of memory and 2 cpus.
Summary
This session has provided the audience with the basics of utilizing Biowulf's compute resources. The audience should now be able to work interactively on Biwoulf's compute nodes or submit batch jobs. The information presented here really just scratched the surface and below are more ways for the audience to learn more or to obtain help.
Biowulf training on YouTube
Interactive sessions Swarm Batch jobs
Biowulf user guide
Biowulf monthly Zoom consults
Biowulf training and Zoom consults
Biowulf batch job coding club: scripts and data
To obtain the scripts and data used for this coding club, sign onto Biowulf and change into the data directory. Then
wget https://bioinformatics.ccr.cancer.gov/docs/btep-coding-club/batch_job_coding_club.zip
mkdir batch_job_coding_club
unzip batch_job_coding_club.zip -d batch_job_coding_club
cd batch_job_coding_club
Everything should be in the /data/username/batch_job_coding_club directory, where username is the user's Biowoulf account ID.