This is part II of the article highlighting nf-core pipelines and specifically addresses the use of these pipelines in the DNAnexus cloud environment. Part I of the article can be found in the October 2023 topic spotlight. As stated previously nf-core is a community effort to generate a curated set of standardized, best-practice, reproducible, documented, NGS analysis pipelines. These pipelines are accessible by CCR researchers on both the NIH HPC system (Biowulf) and the DNAnexus Cloud environment.
DNAnexus is a commercial cloud platform that is available to all CCR researchers via a pilot program sponsored by the Office of Science and Technology Resources (OSTR). Basic information about interacting with the system (creating projects, importing, exporting files, running and monitoring applets) can be found here. The DNAnexus environment contains several prebuilt bioinformatic applications and workflows. Additionally, it is relatively simple to develop new applications and workflows within this environment. However, the focus of this article is the implementation of nf-core pipelines on DNAnexus.
Implementing nf-core pipelines as user friendly applications within the DNAnexus cloud environment has presented some challenges. Importing the pipelines can be readily achieved with the automated procedure provided by DNAnexus. However, the resulting interface is somewhat daunting, due to the way all potential options (>20) are presented, and because not all file inputs (fastqs, genomes and indices) are “point and click”. Many file specifications must be entered manually according to DNAnexus file conventions. In order to make these pipelines accessible to as wide an audience as possible, we have developed a format in which basic pipelines have been wrapped within a pair of programs that greatly simplify the interface. Thus, all pipelines are available in two flavors: 1) the simplified interface with a minimal set of options, pre-indexed genomes, and direct access to the most relevant output; and 2) the raw interface with all options, and all outputs, which while more powerful/flexible, requires more expertise to use effectively.
At the present time, the following nf-core pipelines are available on the DNAnexus system to CCR researchers for the analysis of data from: RNASeq, ChIPSeq, ATACSeq, and CUT&Run experiments. The simplified interface combines ease of use, complete usage documentation, and a compact output that includes IGV session files to allow examination of the generated alignments without having to download the BAM and/or BigWig files.
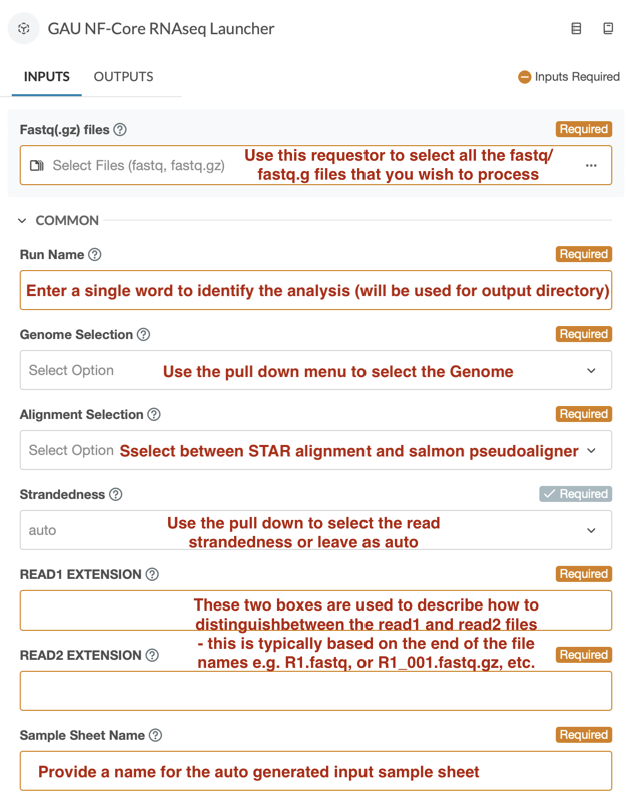
Input form of the simplified interface for the nf-core RNASEQ workflow
To assist potential users in becoming familiar with these applications and their interfaces we are offering a number of introductory classes for each application. The first of these sessions, on RNASeq data analysis will be held on November 16th, 2023 at 1:00 pm. Details and registration information can be found on the BTEP bioinformatic calendar.
Additional classes on other workflows will be forthcoming and will be announced in the near future.
— Peter FitzGerald & Carl McIntosh (Genome Analysis Unit)
This topic spotlight was authored on November 1, 2023 and reflects the information available at that time. As content may evolve, please contact us at ncibtep@nih.gov to obtain the most current information.