Lesson 6: Downloading data from the SRA
For this lesson, you will need to login to the GOLD environment on DNAnexus.
Lesson 5 Review:
- The majority of computational tasks on Biowulf should be submitted as jobs:
sbatchorswarm - the SRA-toolkit can be used to retrieve data from the Sequence Read Archive
Learning Objectives:
- Download data from the SRA with
fastq-dump- split files into forward and reverse reads
- download part, not all, of the data
- Compare
fastq-dumptofasterq-dump - Introduce
prefetch - Look at XML-formatted data with
sra-stat - Grab SRA run info and run accession information
- Work with csv-formatted data using the
cutcommand to isolate columns - Learn to automate data retrieval with the
parallelcommand - Learn about alternative download options
For the remainder of this course, we will be working on the GOLD teaching environment in DNAnexus.
Create a project directory
Today, we will download data from the NCBI/SRA. Let's create a project directory named biostar_class and new folder within biostar_class named sra_data to store the data we are using today. We will use biostar_class for the remaining lessons in this course.
mkdir biostar_class
cd biostar_class
mkdir sra_data
cd sra_data
A brief word about sequence formats
- FASTA – commonly used text format for downstream analysis such as sequence similarity searches
- FASTQ – output format from NGS technologies with quality scores
What is the SRA?
The SRA (Sequence Read Archive) at NCBI is a large, public database of DNA sequencing data. The repository holds "short reads" generated by high-throughput next-generation sequencing, usually less than 1,000 bp.
We will download data from the SRA using the command line package sratoolkit. Note, you can also download files directly from NCBI via a web browser.
Helpful documentation for the SRA:
1. SRA handbook at NCBI
2. SRA Toolkit Documentation (NCBI)
SRA terms to know
NCBI BioProject: PRJN#### (example: PRJNA257197) contains the overall description of a single research initiative; a project will typically relate to multiple samples and datasets.
NCBI BioSample: SAMN#### or SRS#### (example: SAMN03254300) describe biological source material; each physically unique specimen is registered as a single BioSample with a unique set of attributes.
SRA Experiment: SRX#### a unique sequencing library for a specific sample
SRA Run: SRR#### or ERR#### (example: SRR1553610) is a manifest of data file(s) linked to a given sequencing library (experiment). --- Biostar handbook (XIII SEQUENCING DATA)
Using fastq-dump
fastq-dump and fasterq-dump can be used to download FASTQ-formatted data. Both download the data in SRA format and convert it to FASTQ format.
fastq-dump SRR1553607
SRR1553607.fastq
What are "spots"?
Spots are a legacy term referring to locations on the flow cell for Illumina sequencers. All of the bases for a single location constitute the spot including technical reads (e.g., adapters, primers, barcodes, etc.) and biological reads (forward, reverse). In general, you can think of a spot as you do a read. For more information on spots, see the linked discussion on Biostars. When downloading "spots", always split the spots into the original files using:
--split-files
For paired-end reads, we need to separate the data into two different files like this:
fastq-dump --split-files SRR1553607
which creates
SRR1553607_1.fastq
SRR1553607_2.fastq
NOTE: There is an additional option --split-3 that will split the reads into forward and reverse files and a third file with unmatched reads. Since many bioinformatic programs require matching paired end reads, this is the preferred option.
fastq-dump first downloads the data in SRA format, then converts it to FASTQ. If we want to work with a subset of the data, for example the first 10,000 reads, we can use -X:
fastq-dump --split-files -X 10000 SRR1553607 --outdir SRR1553607_subset
We have additionally included --outdir to save our subsetted data to a different directory, so that we do not overwrite our previous downloads.
To see other fastq-dump options, use
fastq-dump --help
Other options of interest:
--gzip or bzip2 allows you to compress the reads
--skip-technical allows you to only download biological reads
To generate an XML report on the data that shows us the "spot" or read count, and the count of bases "base_count", including the size of the data file:
sra-stat --xml --quick SRR1553607
<Run accession="SRR1553607" spot_count="203445" base_count="41095890" base_count_bio="41095890" cmp_base_count="41095890">
<Member member_name="A" spot_count="203445" base_count="41095890" base_count_bio="41095890" cmp_base_count="41095890" library="NM042.2_r1" sample="NM042.2_r1"/>
<Size value="25452196" units="bytes"/>
<AlignInfo>
</AlignInfo>
<QualityCount>
<Quality value="2" count="3523771"/>
<Quality value="5" count="40006"/>
<Quality value="6" count="30160"/>
<Quality value="7" count="79961"/>
<Quality value="8" count="80987"/>
<Quality value="9" count="21904"/>
<Quality value="10" count="63700"/>
<Quality value="11" count="23695"/>
<Quality value="12" count="42479"/>
<Quality value="13" count="24910"/>
<Quality value="14" count="22036"/>
<Quality value="15" count="135747"/>
<Quality value="16" count="56174"/>
<Quality value="17" count="81514"/>
<Quality value="18" count="123393"/>
<Quality value="19" count="79256"/>
<Quality value="20" count="269807"/>
<Quality value="21" count="68295"/>
<Quality value="22" count="125196"/>
<Quality value="23" count="267733"/>
<Quality value="24" count="287688"/>
<Quality value="25" count="296721"/>
<Quality value="26" count="338062"/>
<Quality value="27" count="507890"/>
<Quality value="28" count="226423"/>
<Quality value="29" count="569612"/>
<Quality value="30" count="813014"/>
<Quality value="31" count="1309505"/>
<Quality value="32" count="812951"/>
<Quality value="33" count="2398417"/>
<Quality value="34" count="2324453"/>
<Quality value="35" count="10399039"/>
<Quality value="36" count="1250313"/>
<Quality value="37" count="2681177"/>
<Quality value="38" count="1210457"/>
<Quality value="39" count="2744683"/>
<Quality value="40" count="2268539"/>
<Quality value="41" count="5496222"/>
</QualityCount>
<Databases>
<Database>
<Table name="SEQUENCE">
<Statistics source="meta">
<Rows count="203445"/>
<Elements count="41095890"/>
</Statistics>
</Table>
</Database>
</Databases>
</Run>
spot_count="203445" base_count="41095890"
Using seqkit stat
We can get similar information from our downloaded files using a program called seqkit.
First, let's see what options are available?
seqkit --help
We can see that seqkit stats provides "simple statistics of FASTA/Q files". Let's try it out.
seqkit stat SRR1553607_1.fastq
fasterq-dump
We have already seen fasterq-dump in action (See Lesson 5). fasterq-dump is faster than fastq-dump because it uses multi-threading (default --threads 6). --split-3 and --skip-technical are defaults with fasterq-dump, so you do not need to worry about specifying how you want the files to be split.
However, you can not grab a subset of the file like you can with fastq-dump nor can you compress the file during download, so fasterq-dump is not necessarily a replacement for fastq-dump.
Using prefetch
Both fastq-dump and fasterq-dump are faster when following prefetch, and fasterq-dump paired with prefetch is the fastest way to pull the files from the SRA. There are alternative options, which we will mention later.
prefetch will first download all of the necessary files to your working directory. Runs are downloaded in the SRA format (compressed).
mkdir prefetch
cd prefetch
prefetch SRR1553607
ls -l
fasterq-dump will then convert the files to the fastq format.
fasterq-dump SRR1553607
Navigating NCBI SRA
Where can we get an accession list or run information for a particular project?
Navigating the NCBI website can be challenging. From a user perspective, nothing is as straight forward as you would expect. For the sequence read archive (SRA), there are fortunately some options. There are the convuluted e-utilites, which can grab run information without navigating to the NCBI website, or there is the SRA Run Selector. The Run Selector is nice because you can filter the results for a given BioProject and obtain only the accessions that interest you in a semi user-friendly format. We can search directly from the SRA Run Selector, or simply go to the main NCBI website and begin our search from there. Let's see the latter example, as this is likely the primary way you will attempt to find data in the future.
Step 1: Start from the NCBI homepage. Type the BioProject ID (PRJNA257197) into the search field.
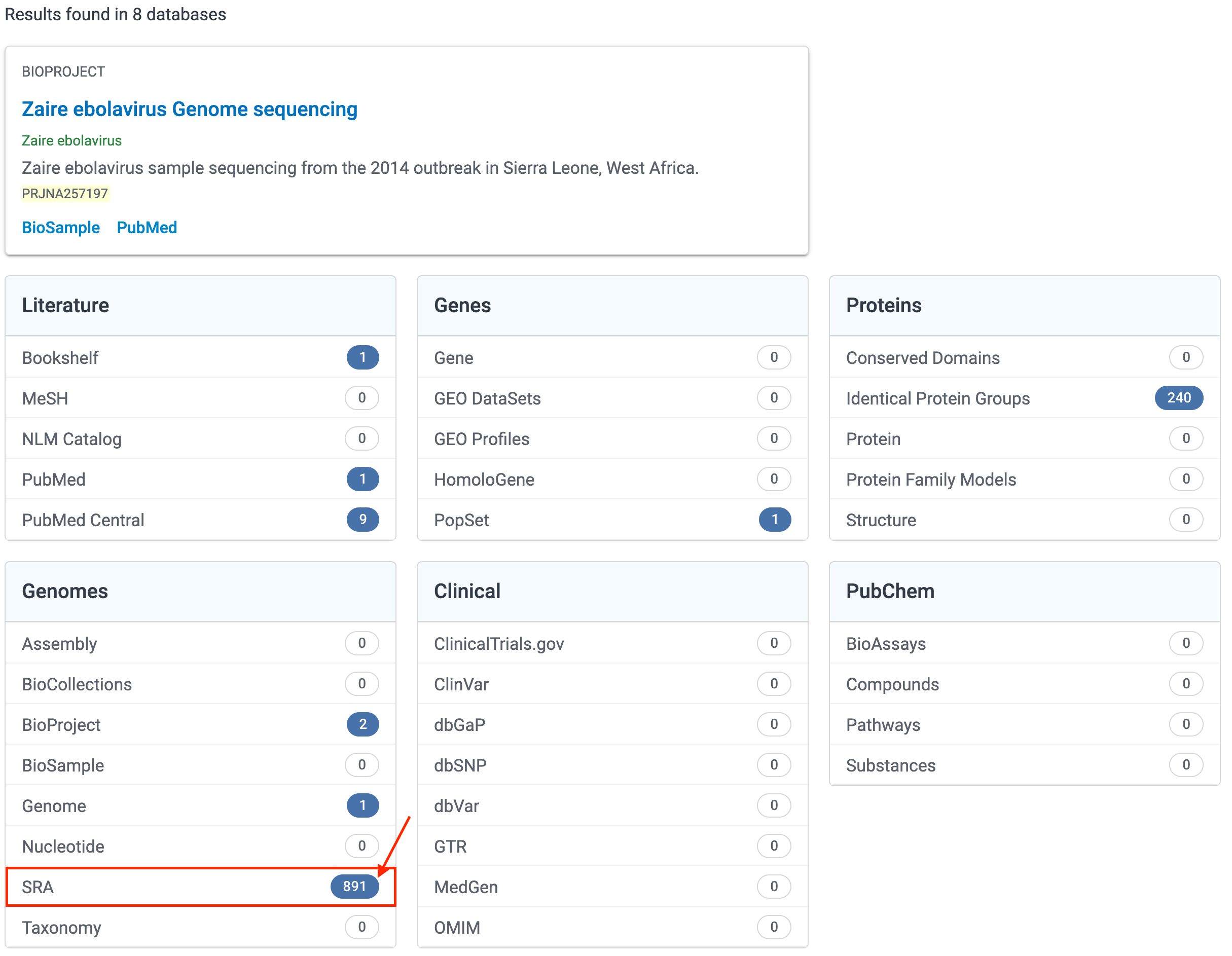
Step 2: In the search results, select the entries next to the SRA.
Step 3: From the SRA results, select "Send results to Run Selector".
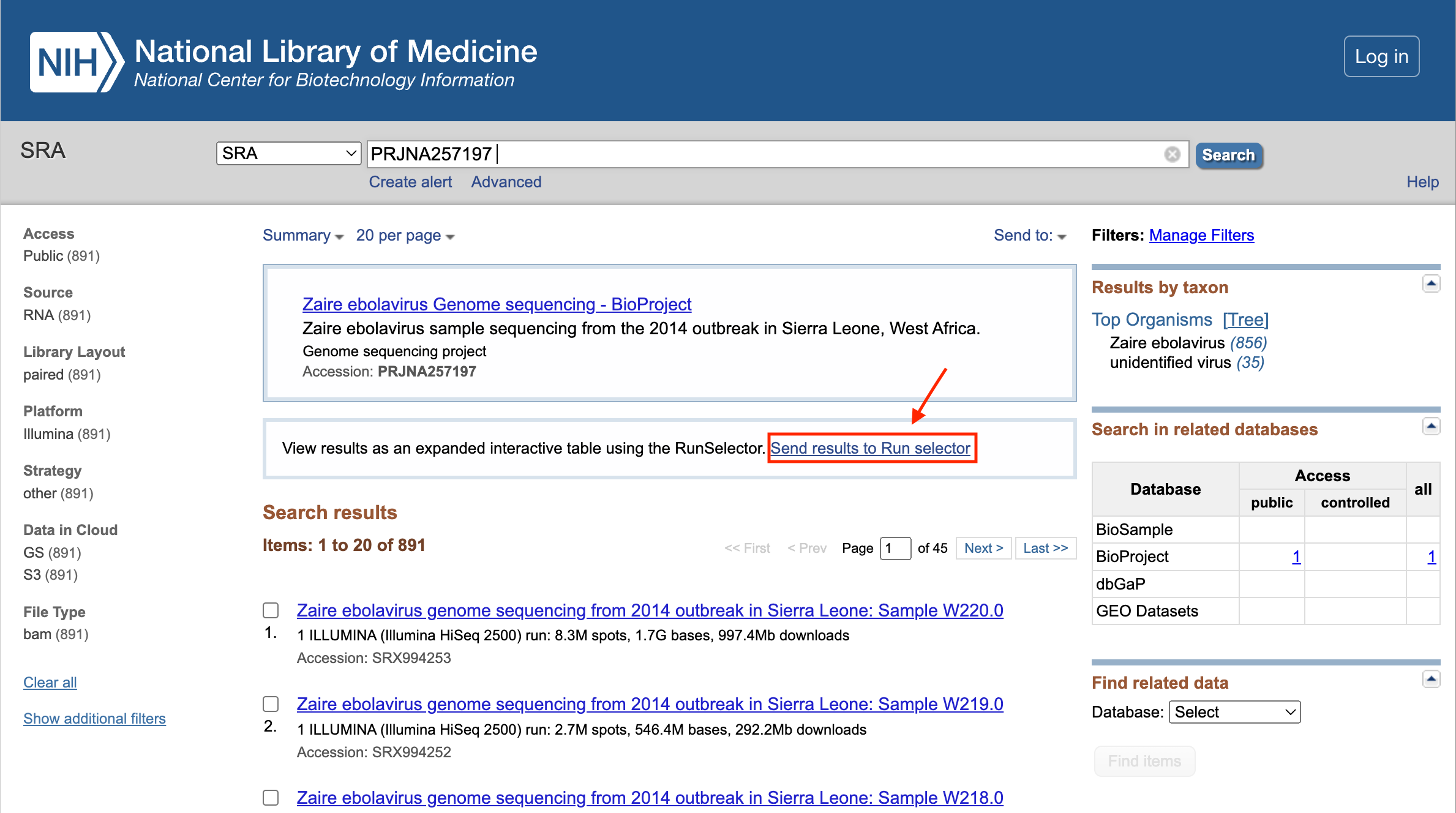
Step 4: From there, you can download the metadata or accession list.
Copy and paste the accession list into a file using nano. Save to runaccessions.txt. Now use head to grab the first few results.
head -n 3 runaccessions.txt > runids.txt
E-utilities
Using e-utilities to programmatically grab the run info
esearch and efetch, can be used to query and pull information from Entrez. They aren't the easiest to understand or use, but you can use them to get the same run info that we grabbed using the Run Selector with the following one liner:
esearch -db sra -query PRJNA257197 | efetch -format runinfo > runinfo.csv
esearch, which queries the SRA. That info can be piped to efetch, which will pull down the run info in comma separated format, which we can save to file using >.
Then we can use a combo of cat, cut, grep, and head to grab only the accession info we are interested in:
cat runinfo.csv | cut -f 1 -d ',' | grep SRR | head > runids.txt
Here, we opened the file with cat, piped it to the cut command, piped it to grep for only the accession IDs (skipping the column header), and get only the first few results.
So what is cut? It is a unix command, that is used to cut out selected portions of the file (man cut for more info). The -f option specifies which field to cut (the first field), and -d tells the program which delimiter to use. We know this is a comma-separate value file, so we do -d ',' to specify the comma.
Using the "parallel" tool for automating downloads
Now that we have accession numbers to work with, let's use parallel and fastq-dump to download the data.
GNU parallel executes commands in "parallel", one for each CPU core on your system. It can serve as a nice replacement of the for loop.
See Tool: Gnu Parallel - Parallelize Serial Command Line Programs Without Changing Them
cat runids.txt | parallel -j 1 fastq-dump -X 10000 --split-files {}
This gives us twenty files, two files for each run and 10 runs. It takes the place of the multiple fastq-dump commands we would need to use.
What's up with the curly braces {}?
The curly braces are default behavior for parallel. This is the default replacement string; when empty, it appends inputs. So the following gets the same result.
cat runids.txt | parallel fastq-dump -X 10000 --split-files
The real power of parallel comes when using something between the brackets such as {.}, {/}, and {//}, like this...
nano filelist
/dir/subdir/file.txt
/other/list.csv
/raw/seqs.fastq
Save the file -> Ctrl O, Yes, Ctrl X.
cat filelist | parallel echo {} "without extension" {.}
/dir/subdir/file.txt without extension /dir/subdir/file
/other/list.csv without extension /other/list
/raw/seqs.fastq without extension /raw/seqs
This replaces the extension.
cat filelist | parallel echo {} "without path" {/}
/dir/subdir/file.txt without path file.txt
/other/list.csv without path list.csv
/raw/seqs.fastq without path seqs.fastq
This removes the path.
cat filelist | parallel echo {} "with root" {//}
/dir/subdir/file.txt with root /dir/subdir
/other/list.csv with root /other
/raw/seqs.fastq with root /raw
This keeps only the path.
Note: parallel will default to 1 concurrent job per available CPU. On a shared system like helix with 48 CPUs, where users do run fast(er)q-dump, that can cause an overload. Even when submitting a job on Biowulf, you may not want to run as many concurrent jobs as there are allocated CPUs (e.g. if you ran fasterq-dump with 2 threads and you have 12 CPUs allocated --jobs would be 6). Therefore, it is good practice to always specify the -j flag, which assigns the number of jobs for the parallel command.
European Nucleotide Archive (ENA)
Everything in the SRA is also in the ENA (See PRJNA257197 on ENA). Files in the ENA share the same naming convention as the SRA but are stored directly as gzipped fastq files and bam files. You can easily use wget or curl to pull files directly from the ENA without using the sratoolkit. More on wget and curl in the next lesson. Check out the ENA training modules for more information on downloading data from the ENA, including examples.
There is also a fantastic web service called SRA Explorer that can be used to easily generate code for file downloads from the SRA, if you are looking to download a maximum of 500 samples. Use caution, the SRA-explorer is not associated with the ENA or NCBI, and program support / updates cannot be guaranteed.
Help Session
Let's search for and download some data.