Questions and answers
BTEP Bioinformatics for Beginners (September 13th, 2022 - December 13th, 2022)
Questions and Answers
Below, you will find questions and answers brought up in the course polls for the BTEP Bioinformatics for Beginners course series that took place from September 13th, 2022 to December 13th, 2022.
Question 1: Normalization - when to normalize and what type of normalization
Answer to Question 1: Normalization is always needed in RNA sequencing because this helps to remove technical effects so that we are comparing only the biological differences between experimental conditions. Technical factors include things like batch effects, differences in sequencing depth (library size) between samples, gene length, and library composition. Various techniques that normalized based on library size and gene length were discussed in class. Refer to the documenation on RNA sequencing quantitation.
Other types of normalization method include quantile normalization, which has been found to work well for RNA sequencing. Further, some differential expression analysis packages have their own normalization scheme so all users need to do is to provide the raw integer expression counts.
Question 2: Question regarding normalization sent by a class participant
“Hi-just a followup comment. The experience of the bioinformatics people that my lab works with is that normalization in RNAseq is everything and you are wasting your time if you don’t do it properly. Especially if your treatment leads to in increase in RNA content of the cell-like resting vs activated T cells. The truth is, most everything goes up under these conditions but most software adjusts things such that half genes go up and half go down. In fact most of what is indicated to go down under these conditions are things that just don’t go up as much as other genes. Anyway, that was a long explanation for why understanding how to do this properly is likely to be crucial. In my opinion, anyway. Thanks very much”
Answer to Question 2: All normalization methods (and consequently differential expression methods) contain assumptions about the data you are analyzing, and there is no single statistical method that can completely capture biological phenomena. It's the job of the analyst to be aware of these assumptions and whether they apply to the data you are analyzing. For example, the most commonly used methods in the detection of differential gene expression (e.g. DeSeq2 and edgeR) assume that most genes are not differentially expressed and start with a correction for library size. This assumption might not apply to your experimental setup: If you suspect your treatment would result in differences in in the total amount of signal (RNA yield) between your two samples (e.g. activated vs. resting t-cells, overexpression of cMyc), normalizing by library size wouldn’t be accurate to the biology of your sample since you’d be removing this difference by normalizing by library size. If you want to capture these absolute differences in signal then you would have to modify your experimental approach (e.g. include a spike-in control - see https://journals.asm.org/doi/full/10.1128/MCB.00970-14).
An additional note on normalization: As stated above most common methods of differential gene expression assume most genes are not differentially expressed. When this is true, as in the case of “classical” RNAseq experiments, then the normalization (and indeed analysis methods) make only minor differences. In this example the majority of the identified DEGs are common to the three analysis methods (DeSeq2, edgeR, and limma-voom) and these use slightly different normalizations.
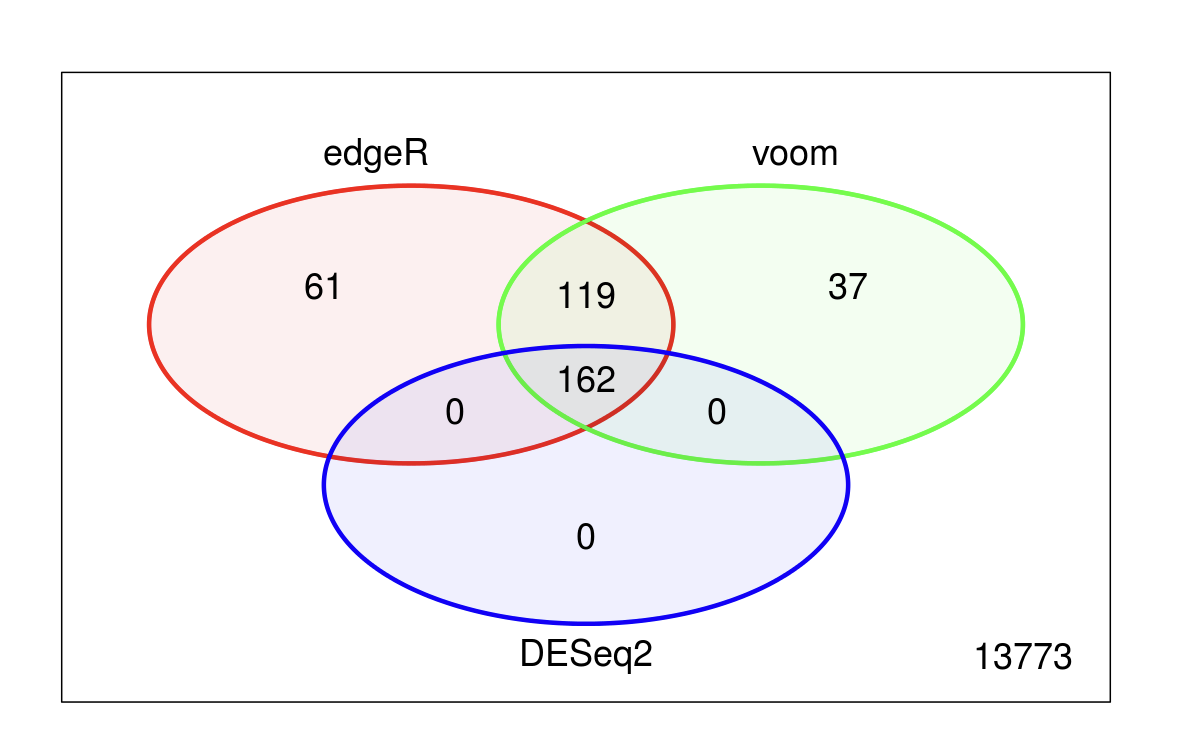
Question 3: Strategies for batch correction (when to use it, what approaches)?
Answer to Question 3: Batch effects are caused by variation between samples that are not due to your experimental design (i.e. technical variation). The best approach to take regarding batch correction is to design your experiments in such a way that you can avoid it entirely: if possible, isolate and prepare your samples on the same day, use the same reagents and locations for preparing your samples). If you must have batches and can’t prepare your samples on the same day, make sure they're not confounded: have representatives of your biological groups (e.g. controls and treatments) in each batch, and keep track of any batches in your experimental records and metadata so you can identify batch effects if they exist in your data (see here for a nice overview).
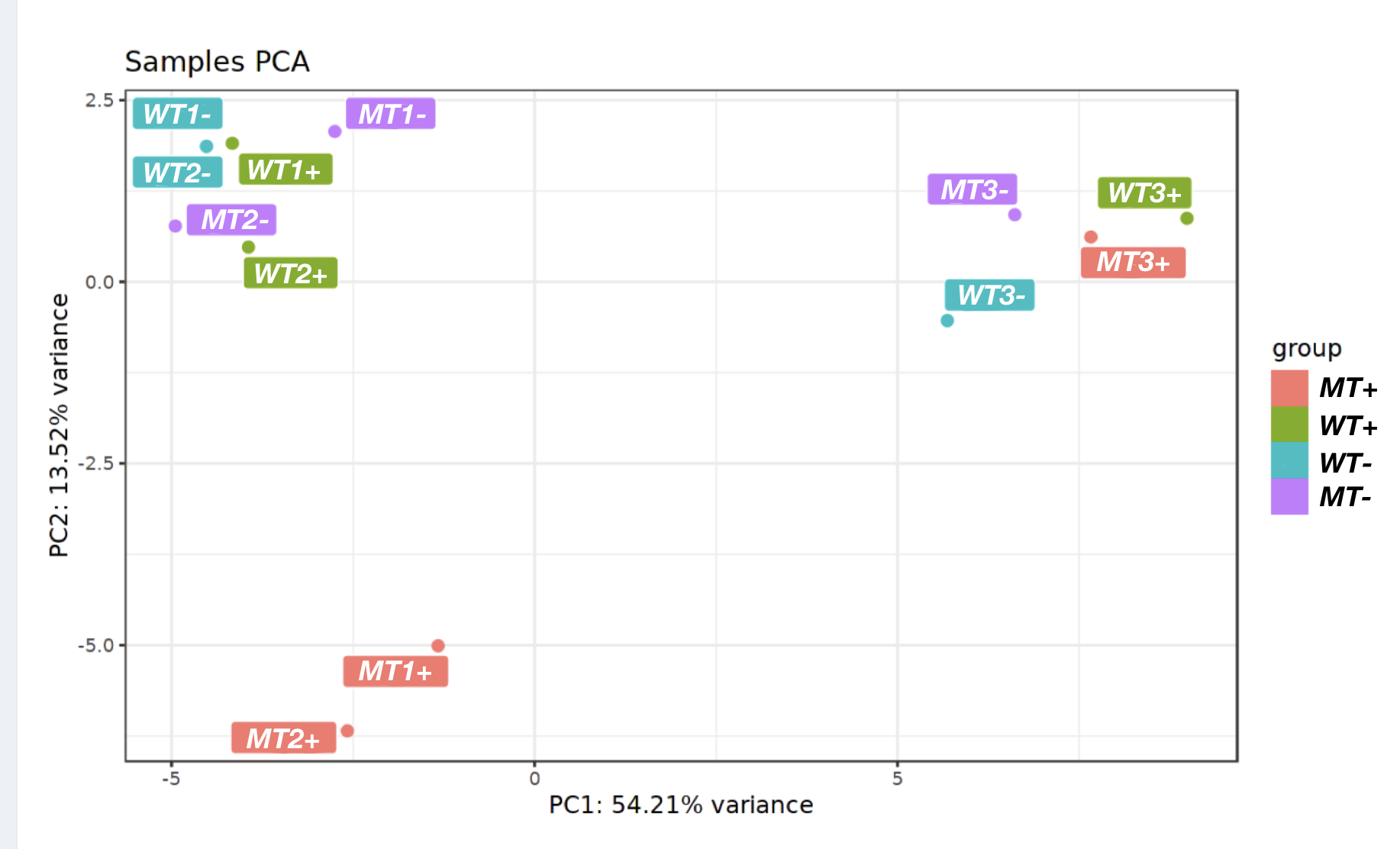
You can identify batch effects through an initial exploratory analysis of your data (e.g. hierarchical clustering, PCA analysis).
An example of how PCA can be used to identify batch effects: When labelled according to sample type (Wildtype +/- drug and Mutant +/- drug) replicates 3 are outliers, and make no sense. However, when knowledge of when the different samples were processed (two “batches”) is added, it is apparent that replicates 3 were all done on a different day and represent batch effects that must be factored into the analysis (and it looks like the drug only affected the mutant).
PCA before batch correction:
PCA after batch correction:
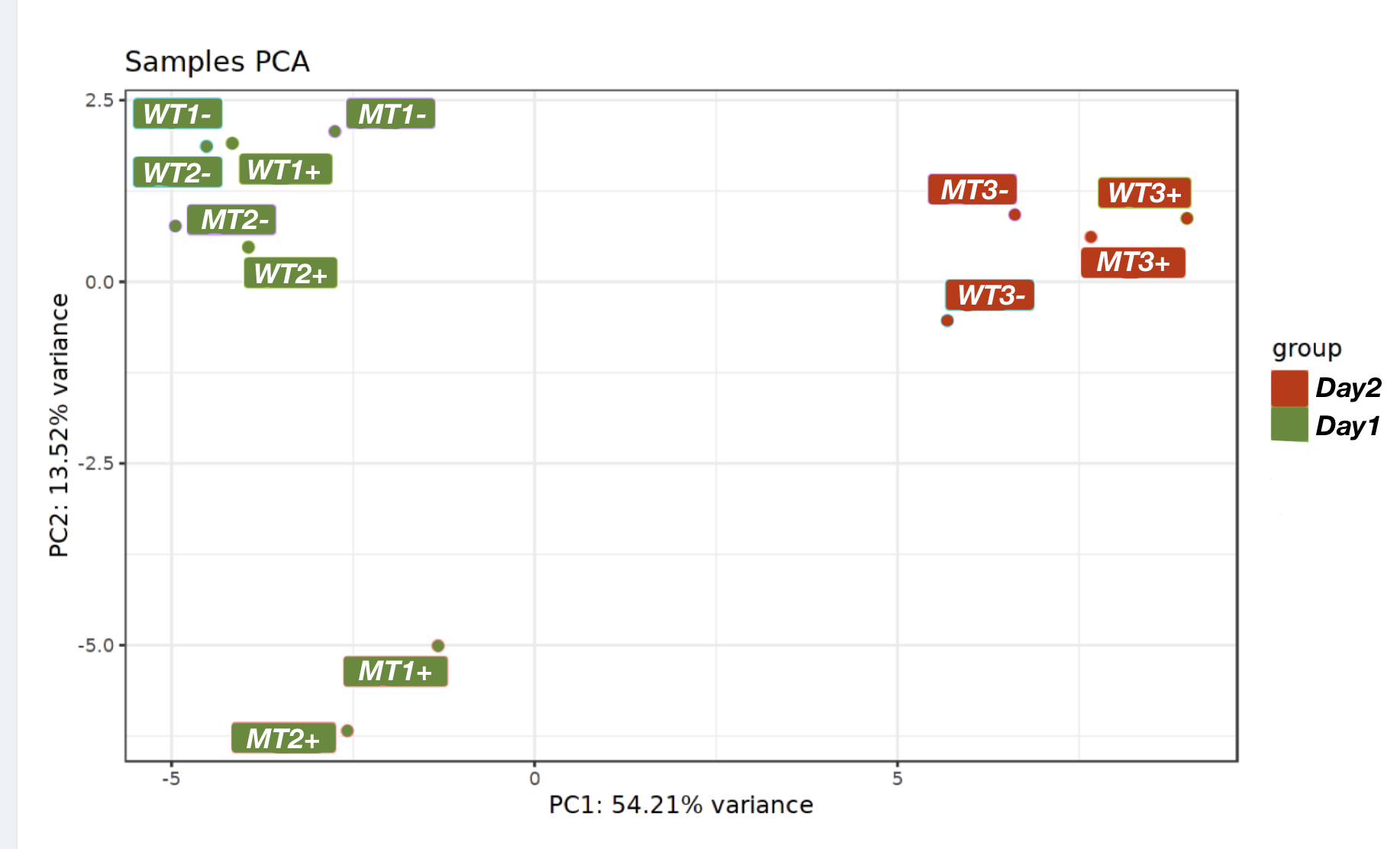
Tools to adjust your data for batch effects exist: there are two common methods, ComBAT and sva which can be found in the Bioconductor package sva. A review on identifying and removing batch effects is reviewed in this article here, however, I would recommend consulting a bioinformatician with experience in this before venturing out to do this on your own.
Question 4: When working with large data sets what is the expectations for time and storage space?
Answer to Question 4: This depends on your project but FASTQ files can be many gigabytes in size and you have FASTQ files for many samples, the space can add up. You can always add more storage space to your Biowulf account. The amount of time it takes to complete your analysis depends on the pipeline that you are using and whether you have to create your own pipeline. If creating your own analysis pipeline, you will also need to optimize the run time of your analysis. Reach out to one of our expert analysts to discuss and plan before starting your study.
Question 5: What methods are available for transferring fastq files from my disk to Biowulf?
Answer to Question 5:
- Mount HPC drives to local machine (slow)
- Use secure copy (scp)
- Use SFTP client (Filezilla, Fugu, or command line SFTP)
- Globus for very large datasets (ie. FASTQ files)
See Transferring data to/from the NIH HPC systems for more about transferring files to and from Biowulf.
Biowulf file transfer tutorials on YouTube
While there are several approaches for transferring file to Biowulf from local, if you are working with many FASTQ files, Globus would be the goto solution.
Question 6: What should I do after downloading RNAseq data from public website(e.g. TCGA) and check for data status?
Answer to Question 6:This depends on the study. For instance, you may find BAM files in a TCGA RNA sequencing study or expression counts. So depending on the data available, this will determine your starting point. When working with public datasets, it is very important to learn as much about a study as you can prior to working with data (ie. do not just download and blindly analyze).
Question 7: What are the various form of data format or quality of public data (GEO, TCGA, Depmap, UK data, etc)?
Answer to Question 7: This varies depending on the repository and study. For instance, GEO does not have a standard set of data that investigators need to submit. Thus, in GEO studies, you may find those with only sequencing data (FASTQ files), expression data, differentially expression results, or a combination.
Question 8: If I want to merge two kinds of GEO study, what should I check in terms of data quality, format, etc?
Answer to Question 8: Start with raw data if at all possible, do some of the QC steps that we did in the class if they’re applicable, and process the data uniformly (same programs, pipelines, etc).