Lesson 4: Useful Unix
For this lesson, you will need to login to the GOLD environment on DNAnexus.
Lesson 3 Review
- Biowulf is the high performance computing cluster at NIH.
- When you apply for a Biowulf account you will be issued two primary storage spaces: 1)
/home/$Userand 2)/data/$USER, with 16 GB and 100 GB of default disk space. - Hundreds of pre-installed bioinformatics programs are available through the
modulesystem. - Computational tasks on Biowulf should be submitted as a job (
sbatch,swarm) or through an interactive sessionsinteractive. - Do not run computational tasks on the login node.
Learning Objectives
We are going to shift gears back to unix. We will focus on learning concepts that make working with unix particularly useful including:
- Flags and command options - making programs do what they do
- Wildcards (
*) - Tab complete for less typing
- Accessing user history with the "up" and "down" arrows on the keyboard
cat,head, andtailfor reading files- Working with file content (input,output, and append)
- Combining commands with the pipe (
|). Where the heck is pipe anyway? - Finding information in files with
grep - Performing repetitive actions with Unix (
for loop) - Permissions - when all else fails check the permissions
Flags and command options - making programs do what they do
Compare the output from these commands:
ls
ls -S
ls -lh
ls -h (when used with -l option, prints file sizes in a human readable format with the unit suffixes: Byte, Kilobyte, Megabyte, Gigabyte, Terabyte. This reduces the number of digits displayed.)
ls -l (list in long format). The column output is as follows:
- file type
- Content permissions
- Number of hard links to content
- Owner
- Group owner
- Content size (bytes)
- Last modified date / time
- File / directory name
-S (sort from largest to smallest file size)
What do you see when combining the -h and -l flags?
There are many flags you can use with ls. How would we find out what they are?
man ls
Or to see a more user friendly display, google to the rescue. Google "man ls unix" and see what you get. Here's a useful, readable explanation of the "ls" command with examples.
Try combining some of the ls flags.
ls -lhS
ls -alt
-a (show hidden dot files .)
-t (sort by modification time with most recent listed first)
Flags and options add a layer of complexity to unix commands but are necessary to get the command or program to behave in the way you expect. For example, here is a command line for running "blastn" an NCBI/BLAST application.
blastn -db nt -query seq1.fasta -out results.out
What's going on in this command line? First, the BLAST algorithm is specified, in this case it is blastn, then the -db flag is used to choose the database to search against (nt for nucleotide). The query flag specifies the input sequence, which is in FASTA format, and the -out flag specifies the name of the output file.
Use of wildcards
Wildcard characters are a handy tool when working at the command line. Want to list all your FASTA files?
Use *:
ls /data/*.fasta
* wildcard matches zero or more characters including spaces.
This example will work as long as all your FASTA files end in .fasta. But sometimes they don't.
ls /data/*.fa
or you want all the the FASTA and FASTQ files.
ls /data/*.f*
In addition to the asterisk (*) character, you can use other wildcards on the unix command line, not limited to the following:
? - matches a single character
{} - used for multiple matches
[] - specify a range of characters or numbers. For example, [a-z] would specify all lower case letters and [0-9] would mean all digits.
To see some more practical examples of using wildcards, see this article from tecmint.com and this from the medium.com. This second article provides a nice discussion on how wildcards differ from regular expressions.
Using tab complete for less typing
Here's a good Unix trick to know - tab complete. Start typing the name of the file or directory you want, and hit the tab key. The system will auto-complete the name of the file or directory if the name is unique. It may only give a partial name if there is more than one file with a similar name in the directory. You can fill in the next part of the name and then try tab-complete again.
Let's create some files to test this.
touch file.txt
touch file.fasta
touch file.fastq
Start typing...
less f (hit tab)
What do you get? How does that differ from:
less file (hit tab)
The tab complete will save you lots of typing, and also help to figure out if you are where you think you are in the directory structure.
Access your history with the "up" and "down" arrows on your keyboard
Here's another Unix trick to make your life easier. Access previous commands with the up and down arrows on your keyboard. You can scroll backwards and forwards. This helps when you've got a typo or small mistake in your command lines that you can fix without retyping the whole thing.
Keyboard shortcuts
There are also a few handy keyboard shortcuts to make life on the command line easier. For example:
ctrl c to kill a running process
ctrl l to clear the screen
ctrl a skip to the beginning of a command
ctrl e skip to the end of a line
See more examples here.
cat, head, and tail
Who says Unix programmers don't have a sense of humor? Let me introduce cat, head, and tail. The cat command (short for "concatenate") is an extremely useful command for creating new files and viewing file contents. You can use it to open files for reading input and writing output. Or you can use it to copy several files into a new file. Also you can "append" the contents of a file to the end of another file.
This command reads the content of sample.fasta and outputs to standard output (i.e., the screen). This is not helpful for very large files, as it moves to the end of the file quickly. Less is a better command option for reading large files.
cat /data/sample.fasta
You can use cat to combine several files into one file, such as:
cat /data/seq1.fasta /data/seq2.fasta
Although, this again prints to standard output, the screen. To capture that output, we need to learn how to redirect output. (Coming up next!)
In the meantime, let's take a quick look at head and tail.
head
head /data/sample.fasta
tail
tail /data/sample.fasta
You can specify how many lines you would like to see (-n), or you can use the default value, which is 10.
head -n 20 /data/sample.fasta
Want to be sure of those 20 lines? cat again.
cat -n /data/sample.fasta
What does the -n flag do? How could you find out more about "cat"?
man cat
Working with file content (input <, and output >, append >>)
<
>
>>
Want to put the output from cat, head, or tail into a new file?
head -n 20 /data/seq1.fasta > smaller.fasta
Or we could put the last 20 lines into a file with tail.
tail -n 20 /data/seq1.fasta > smaller2.fasta
What if we want the first 20 lines and the last 20 lines in one file, with the first at the top and the last at the bottom? Use append, >>
to paste the second file to the bottom of the first file. Let's try it.
head -n 20 /data/sample.fasta > smaller.fasta
tail -n 20 /data/sample.fasta >> smaller.fasta
Keep in mind that if you input into the same file multiple times, you are overwriting the previous contents. For example, what is the final content of our file covid.fasta?
head -n 20 /data/seq1.fasta > covid.fasta
head -n 20 /data/seq2.fasta > covid.fasta
head -n 20 /data/seq3.fasta > covid.fasta
How many lines are now in "covid.fasta"? How can you check?
Let's try wc, short for word count.
wc covid.fasta
wc is a very useful function. Without opening a file, we can find out how many lines, words and characters are in it. Line counts are extremely useful to assess your data output.
If we created a file where we were expecting there to be 1000 lines of output? The wc command provides a quick way to check.
What happened to all of our content? The final results are from "seq3.fasta" only. The other two results files have been overwritten.
So, how would you get all three files into covid.fasta? You'll need to use append.
cat /data/seq1.fasta > covid.fasta
cat /data/seq2.fasta >> covid.fasta
cat /data/seq3.fasta >> covid.fasta
How could you test to see if the file has the expected number of lines?
wc covid.fasta
To input into a file
less < covid.fasta
Combining commands with pipe (|). Where the heck is pipe anyway?
The hardest thing about using pipe (|) is finding it on your keyboard!
The pipe symbol "|" (a.k.a., vertical bar) is way over on the right hand side of your keyboard, above the backslash \.
Pipe is used to take the output from one command, and use it as input for the next command, all in one command line. Let's look at some examples.
head -n 20 /data/sample.fasta | wc
Using what we've learned, all together now.
Let's say you've got a very large FASTA or FASTQ file, and you want to run an analysis on it. Before working on the whole file, it can be useful to set up a smaller test file instead.
Here's one way to do it.
cat /data/sample.fasta | head -n 20 > output.fasta
This combines several things we have learned about. The cat command opens the file sample.fasta for writing. The pipe | command is used to take that output and run it through the head command where we only want to see the first 20 lines, and we want them output > into a file called "output.fasta". Let's compare the files. How are they different?
ls -lh
and
less /data/sample.fasta
less output.fasta
Finding information in files with grep
The grep utility is used to search files looking for a pattern match. It is used like this.
grep pattern options filename
As our first example we will look for restriction enzyme (EcoRI) sites in a sequence file (eco.fasta). The file has four EcoRI sites, but two of them are across the end of the line (and won't be found).
ls /data/eco.fasta
grep -n GAATTC /data/eco.fasta
We can modify the "eco.fasta" file to remove the line breaks (\n) at the ends of the lines.
grep -v ">" /data/eco.fasta | tr -d "\n" | grep GAATTC
-v : This prints out all the lines that do not match the pattern
The unix "tr" (translate) command is used for translating or deleting characters.
Usage:
tr [option] set1 [set2]
-d : Deletes characters in the first set from the output
So this part of the command line is finding the line breaks "\n" and removing them.
The header lines (>) can also be removed.
And now we can see all four of the EcoRI sites.
What if we just wanted to count the occurrence of the EcoRI sites in the sequence?
grep -v ">" /data/eco.fasta | tr -d "\n" | grep GAATTC -c
-c : This prints only a count of the lines that match a pattern.
It is counting the entire line as one.
If we wanted to see each of the EcoRI sites listed.
grep -v ">" /data/eco.fasta | tr -d "\n" | grep GAATTC -o
And if we want to count the number of EcoRI sites.
grep -v ">" /data/eco.fasta | tr -d "\n" | grep GAATTC -o | wc
Let's create a word file that we can input to grep. We can input multiple restriction enzyme sites and search for all of them.
cd
nano wordfile.txt
Put in the words (GAATTC, TTTTT). Now we can use that file to find lines.
grep -v ">" /data/eco.fasta | tr -d "\n" | grep -f wordfile.txt -o | wc
We can find a list of options to be used with grep using man:
man grep
There are also existing programs to find motifs (patterns) in sequence data. For example:
Emboss - fuzznuc - a motif finding program
fuzznuc -help
fuzznuc -pattern GAATTC -rformat2 tagseq eco.fasta -outfile /home/username/results
-rformat2 : Report format
tagseq : tag sequence
-outfile : name and path to the results file
Performing repetitive actions with Unix
We can create a "for" loop to do iterative actions in Unix.
For each commands all on one line or separate lines: (“i” can be any variable name). These steps can be saved as a file, thereby creating a simple Unix script.
What does this "for loop" do?
for i in /data/*.fasta; do ls $i; done
What do these command lines do?
This one pulls out all the header ">" lines in the fasta files.
for i in /data/*.fasta; do echo $i; grep ">" $i; done
While this one just pulls out the ones from files named seq*.fasta.
for i in /data/seq*.fasta; do echo $i; grep ">" $i; done
Permissions - when all else fails check the permissions
Permissions dictate who can access your files and directories, and what actions they can perform. If you are consistently getting error messages from your command line and you're sure you are typing it correctly, it's worthwhile checking the permissions. So what does it all mean? How do we read the permissions information?
As we have seen, ls -l, provides information about file types, the owner of the file, and other permissions.
For example:
ls -l /data/sample.fasta
Here is an overview of what these permissions mean:
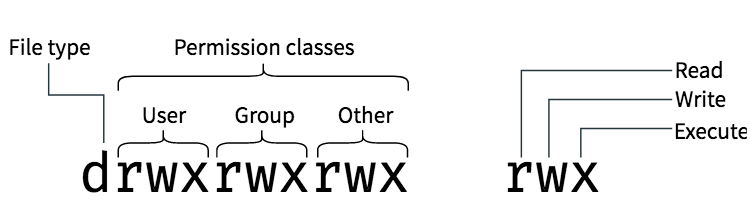
Image from booleanworld.com
The first letter d indicates whether this is a directory or not - or some other special file type. The next 3 positions are the owner's/user's permissions. In this image, the owner can "read", "write" and "execute". So they can create files and directories here, read files here, and execute/run programs. The next 3 positions show the permissions for the "group". The last 3 positions shows permissions for everyone ("other").
You can modify permissions using chmod. Let's see this in action.
touch example.txt
chmod u-w example.txt
ls -l example.txt
chmod u+w example.txt
ls -l example.txt
Help Session
Let's complete a Unix treasure hunt.