RNA-SEQ Overview
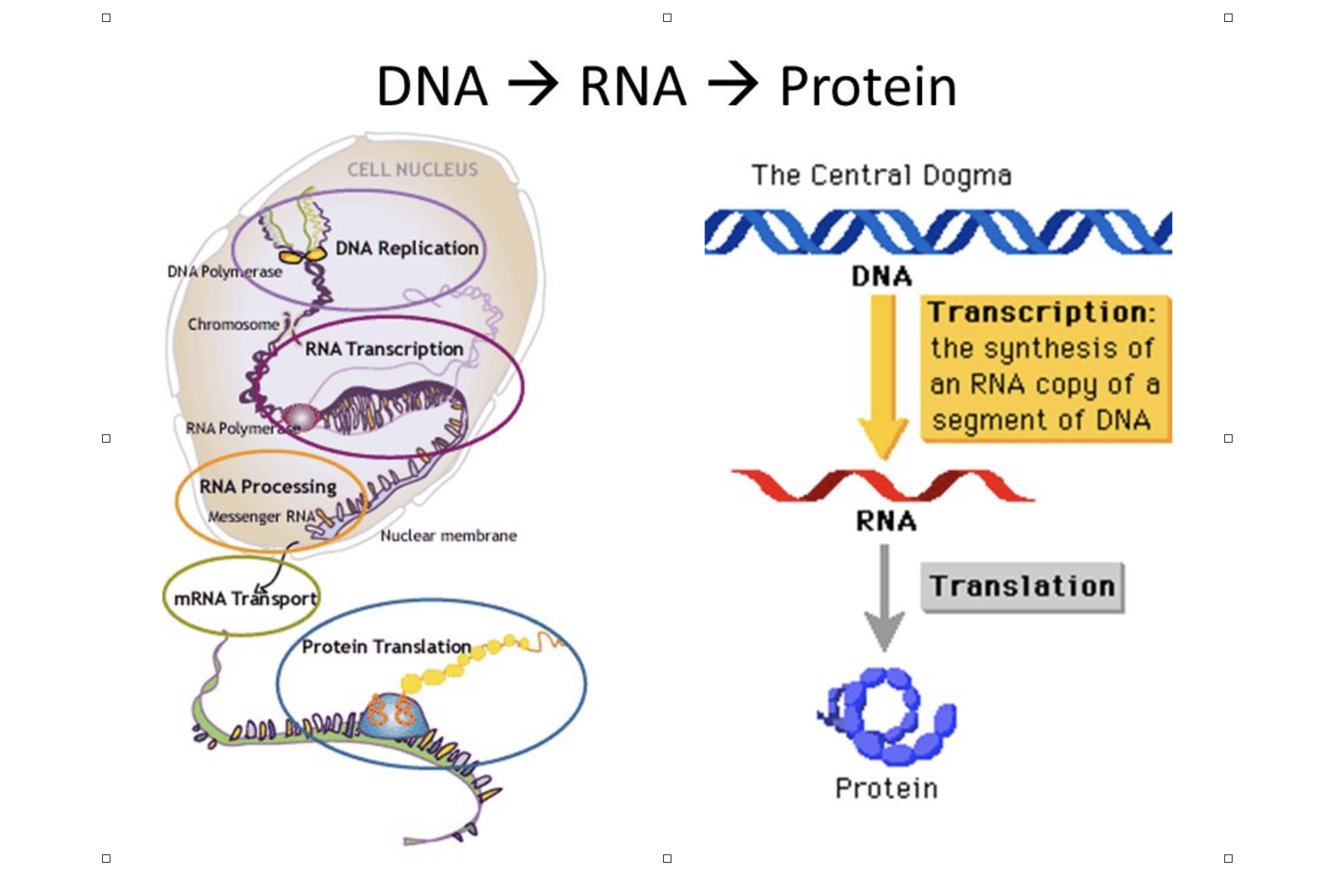
What is RNASEQ ?
RNA-Seq (RNA sequencing), uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment. (Wikipedia) Strictly speaking this could be any type of RNA (mRNA, rRNA, tRNA, snoRNA, miRNA) from any type of biological sample. For the purpose of this talk we will be limiting ourselves to mRNA.
Technically, with a few exceptions, we are not actually sequencing mRNA but rather cDNA.
(RNASeq is only valid within the context of Differential Expression)
RNASEQ - WorkFlow
A typical RNASEQ experiment involves several steps, only one of which falls within the realm of bioinformatics. Namely the Data Analysis step.
- Experimental Design
- What question am I asking
- How should I do it (does it need to be done)
- Sample Preparation
- Sample Prep
- Library Prep
- Quality Assurance
- Sequencing
- Technology/Platform
- Detail Choices
- Data Analysis (Computation)
- Quality Contol
- Alignment
- Quantitation
- Differential Expression
- Biological Meaning
We will now examine each of this steps, highlight the major components of each, and touching briefly on some of more critical steps and pitfalls.
Generating the Data - Experimental Design
A good experimental design is vital for the success of any RNASEQ experiment. Before you begin the experiment make sure you have a clear understanding of the technique and how to avoid costly mistakes that can produce unanalysable data. Listed below are some of the things you should consider before starting your experiment
Only Sequence the RNA of interest
- Remember ~90% of RNA is ribosomal RNA. Therefore enrich your total RNA sample by: polyA selection (oligodT affinity) of mRNA (eukaryote), or rRNA depletion - RiboZero is typically used (costs extra)
Remember
- RNASEQ looks at steady state mRNA levels which is the sum of transcription and degradation
- Protein levels are assumed to be driven by mRNA levels
- RNASEQ can measure relative abundance not absolute abundance
- RNASEQ is really all about sequencing cDNA
What question(s) are you asking?
Your answers to the following questions will dictate many of your choices with respect of the appropriate methodologies to be employed.
- Which gene are expressed?
- Which genes are differentially expressed?
- Are different splicing isoforms expressed?
- Are there novel genes or isoforms expressed?
- Are you interested in structural variants or SNPs, indels Are you interested in non-coding RNAs
- Does your interest lie in micro RNAs
- If this a standalone experiment, a pilot, or a “fishing trip”
Read Choices
For any NGS experiment you will have to make choices about the following sequencing options. Unfortunately, there is and inverse relationship between accuracy and cost.
- Read Depth
- More depth needed for lowly expressed genes
- Detecting low fold differences need more depth
- Read Length
- The longer the length the more likely to map uniquely
- Paired read help in mapping and junctions
- Replicates
- Detecting subtle differences in expression needs more replicates
- Detecting novel genes or alternate iso-forms need more replicates
*Increasing depth, length, and/or replicates increase costs
Replicates
Technical Replicates
- It’s generally accepted that they are not necessary because of the low technical variation in RNASeq experiments
Biological Replicates (Always useful)
- Not strictly needed for the identification of novel transcripts and transcriptome assembly.
- Essential for differential expression analysis - must have 3+ for statistical analysis
- Minimum number of replicates needed is variable and difficult to determine:
- 3+ for cell lines
- 5+ for inbred samples
- 20+ for human samples (rarely possible)
- More is always better
Data Analysis Questions
Make sure you have a clear plan for storing, managing and analysing your data. Also, ensure you have a method to capture all the pertinant metadata and document the data analysis steps that have been taken.
- Where will the primary data be stored (fastq)? Where will the processed data be stored (bam)? Who will do the primary analysis?
- Who will do the secondary analysis?
- Where will the published data be deposited and by who? (what metadata will they require)
- Are you doing reproducible science?
If you are not going to analyse the data yourself talk to the people who will be analyzing your data BEFORE doing the experiment*
Best Practice Guidelines from Bioinformatic Core (CCBR):
- Factor in at least 3 replicates (absolute minimum), but 4 if possible (optimum minimum). Biological replicates are recommended rather than technical replicates.
- Always process your RNA extractions at the same time. Extractions done at different times lead to unwanted batch effects.
- There are 2 major considerations for RNA-Seq libraries: • If you are interested in coding mRNA, you can select to use the mRNA library prep. The recommended sequencing depth is between 10-20M paired-end (PE) reads. Your RNA has to be high quality (RIN > 8). • If you are interested in long noncoding RNA as well, you can select the total RNA method, with sequencing depth ~25-60M PE reads. This is also an option if your RNA is degraded.
- Ideally to avoid lane batch effects, all samples would need to be multiplexed together and run on the same lane. This may require an initial MiSeq run for library balancing. Additional lanes can be run if more sequencing depth is needed.
- If you are unable to process all your RNA samples together and need to process them in batches, make sure that replicates for each condition are in each batch so that the batch effects can be measured and removed bioinformatically.
- For sequence depth and machine requirements, visit Illumina Sequencing Coverage website
For cost estimates, visit Sequencing Facility pricing for NGS For further assistance in planning your RNA-Seq experiment or to discuss specifics of your project, please contact us by email: CCBR@mail.nih.gov OR visit us during office hours on Fridays 10am to noon (Bldg37/Room3041). For cost and specific information about setting up an RNA-Seq experiment, please visit the Sequencing Facility website or contact Bao Tran