Quantitation
Counting as a measure of Expression
Most RNASEQ techniques deal with count data.
- The reads are mapped to a reference and the number of reads mapped to each gene/transcript is counted
- Read counts are roughly proportional to gene-length and abundance
- The more reads the better
- Artifacts occur because of:
- Sequencing Bias
- Positional bias along the length of the gene Gene annotations (overlapping genes) Alternate splicing
- Non-unique genes
- Mapping errors
- Artifacts occur because of:
The typical steps in quantitation of mapped reads is as follows:
- Count mapped reads
- Count each read once (deduplicate)
- Discard reads that:
- have poor quality alignment scores
- are not uniquely mapped
- overlap several genes
- Have paired reads do not map together
- Remember to document what was done
Count Normalization

There are three metrics commonly used to attempt to normalize for sequencing depth and gene length.

Counting as a measure of Expression
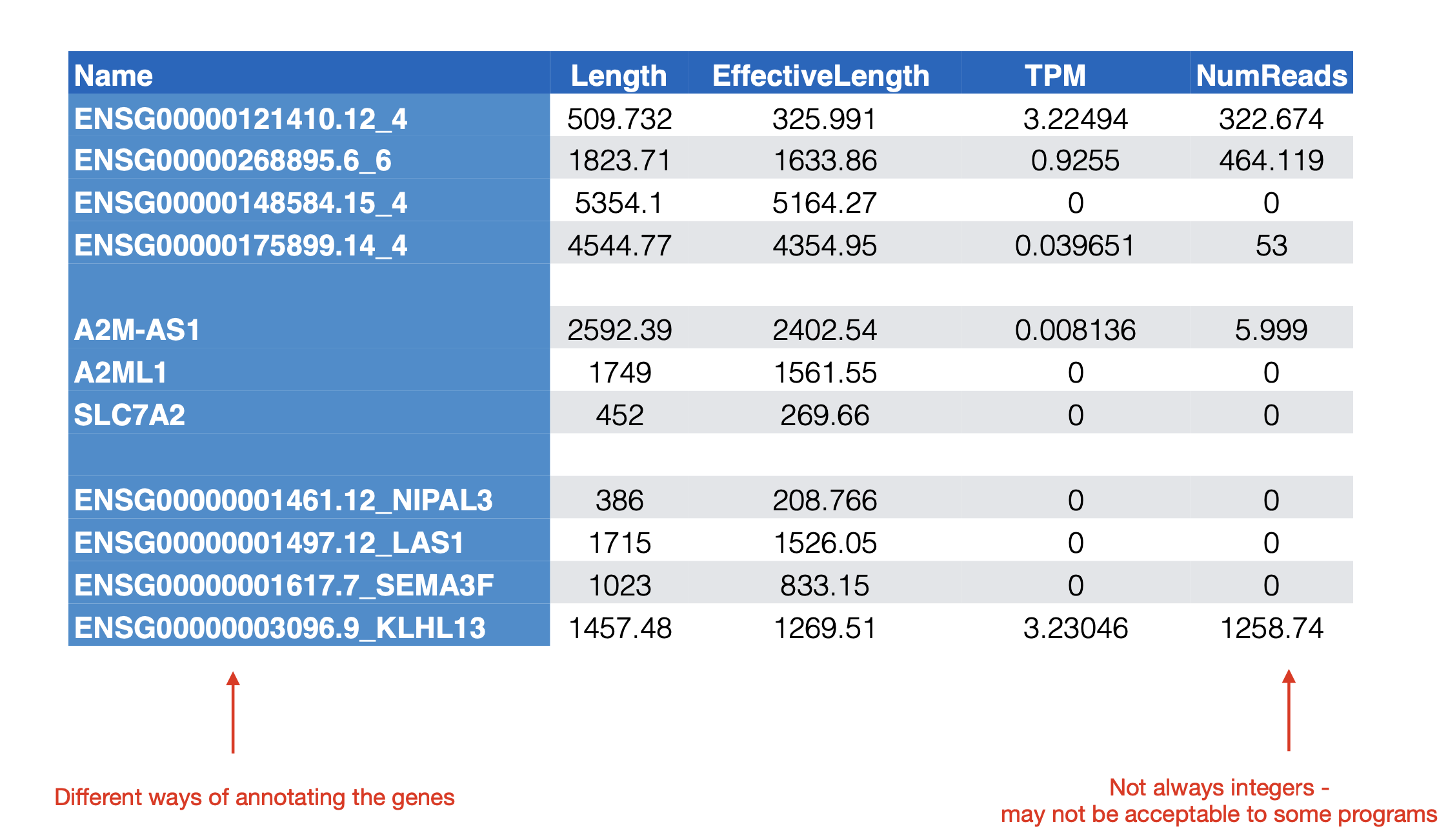
An example of a counts matrix for RNASEQ data.
Log Transformed Data
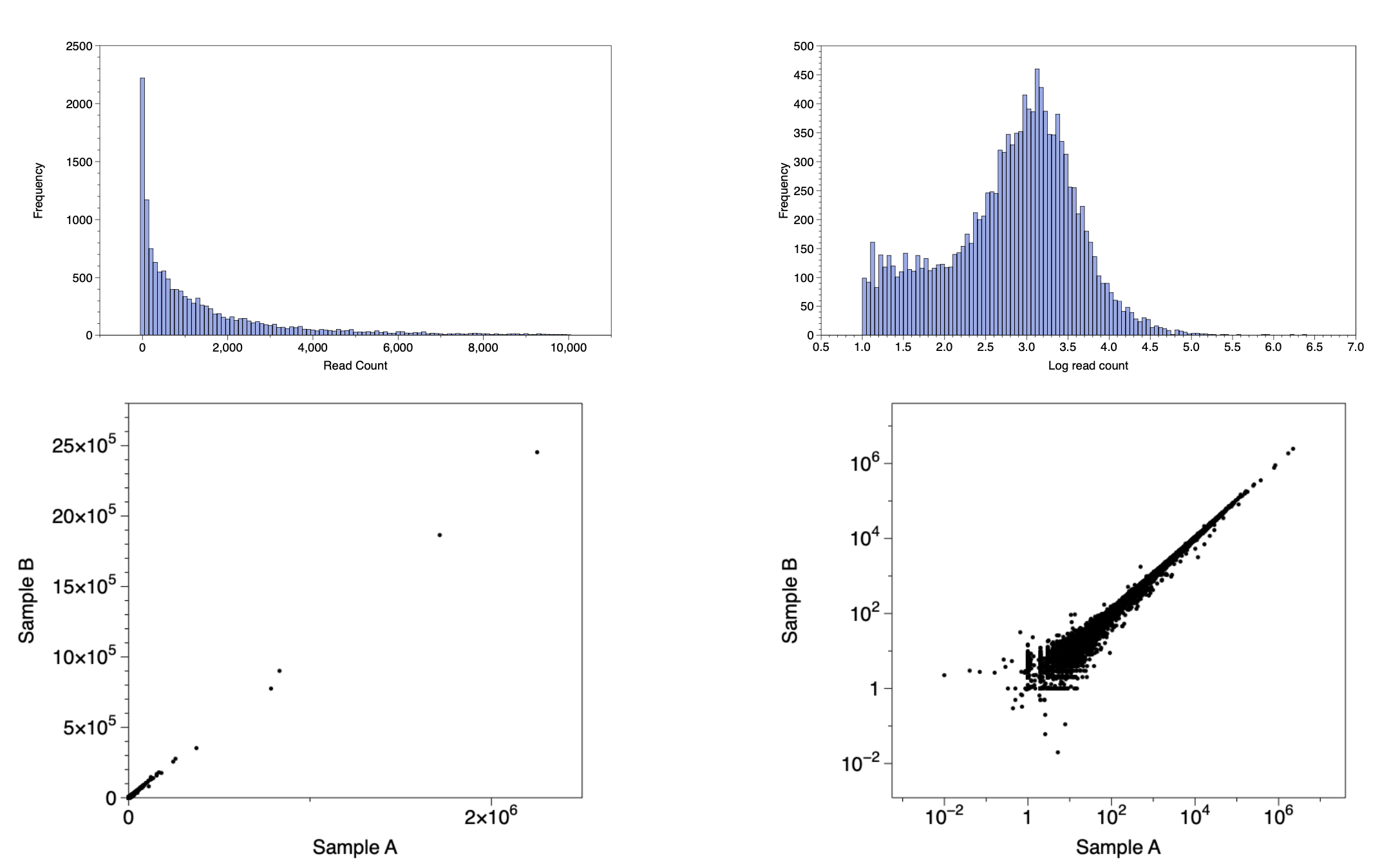
Because of its vast dynamic ranges RNASEQ data is typically log transformed in order to: provide better visualizations and to present analysis software with a more "normal distribution".
Common counting Programs
- Subread (featureCount)
- STAR (quantmode)
- HTseq (counts)
- RSEM (RNA-Seq by Expectation Maximization) Salmon, Kallisto - pseudoaligners
- Salmon (pseudo aligner and counter)