Lesson 13: Aligning raw sequences to reference genome
Before getting started, remember to be signed on to the DNAnexus GOLD environment.
Lesson 11 Review
In Lesson 11 we learned to aggregate multiple FASTQC reports into one using MultiQC, which allows us to easily interrogate the quality of sequencing data for multiple samples. We also learned to trim away adapters and poor quality reads in raw data using Trimmomatic (instructions for trimming using BBDuk are available in the Lesson 11 content for you to look at even though we did not go over this).
Learning objectives
In this lesson, we will continue to use the Human Brain Reference (HBR) and Universal Human Reference (UHR) data and we will
- Learn to align the sequencing data to reference genome using HISAT2, which is a splice aware aligner
- Look at post alignment QC
- Familiarize ourselves with the contents of alignment output
- Learn to use SAMTOOLS to work with alignment output
- Align sequences with BOWTIE2, which is not splice aware so we can visualize and compare to results obtained from HISAT2 using the Integrative Genome Viewer (IGV) in Lesson 14.
The skills learned in this lesson can be applied towards your own research and subsequent lessons in this course.
Creating an index for the reference genome
Let's start our exploration of sequencing read alignment by discussing the reference genome for human chromosome 22. For this, change into our ~/biostar_class/hbr_uhr/refs folder. In Lesson 9, we discussed why we need a reference genome when we are working with high throughput sequencing data. In high throughput sequencing, we obtain many sequence reads in our experimental output and we do not know where in the genome these reads come from. The reference genome is a known standard that helps us reassemble these reads.
cd ~/biostar_class/hbr_uhr/refs
As a review, when we list the contents of this folder, we will see that it contains the reference genome (22.fa) and annotation (22.gtf) for human chromosome 22.
ls
22.fa 22.gtf ERCC92.fa ERCC92.gtf
The first step in alignment is to create an index for the reference genome. Think of an index as a table of contents in a book. If we are searching for something in a book, we can either search from beginning to end and depending on the size of the book, this could take a long time. Alternatively, we could use the table of contents to jump to and search only the relevant sections. Thus, an index allows the aligner to search more specifically and reduce computation time.
To align the HBR and UHR raw reads to chromosome 22 we will use a tool called HISAT2, which is a splice aware aligner used for RNA sequencing. This aligner will be able to handle the alignment of reads that fall on two exons. We will use the build feature of HISAT2 to create our index. Other aligners will have their own algorithm for indexing the reference genome.
To build the index for human chromosome 22, we type hisat2-build at the command prompt, followed by the name of FASTA file for the reference genome (22.fa in our case) and then the base name (ie. file name without extension) that we would like to use for our index (here we choose 22 as the base name).
hisat2-build 22.fa 22
After the index has been generated, we can list the contents of our biostar_class/hbr_uhr/refs folder to see if anything has changed. We use the -1 option with ls to list one item per row.
ls -1
Note that we now have HISAT2 genome indices, which are the 8 files that have extension "ht2".
22.1.ht2
22.2.ht2
22.3.ht2
22.4.ht2
22.5.ht2
22.6.ht2
22.7.ht2
22.8.ht2
Once the index for the human chromosome 22 reference has been created, change back into the ~/biostar_class/hbr_uhr folder.
cd ~/biostar_class/hbr_uhr
Then create a new folder called hbr_uhr_hisat2 and change into this. We will keep our alignment outputs in this folder.
mkdir hbr_uhr_hisat2
cd hbr_uhr_hisat2
Performing alignment for one file using HISAT2
To align FASTQ files for one sample, we construct the HISAT2 command with the following options.
- The "-x" flag prompts us to enter the base name (ie. without extension) of genome index. The HISAT2 index in the ~/biostar_class/hbr_uhr/refs directory, which is one directory back from our present working directory of ~/biostar_class/hbr_uhr/hbr_uhr_hisat2, so we can use ".." to specify go one directory back then go into refs.
- We specify files containing read 1 and read 2 of paired end sequencing after "-1" and "-2" flags, respectively. The reads are in ~/biostar_class/hbr_uhr/reads, which is one directory back from our present working directory of ~/biostar_class/hbr_uhr/hbr_uhr_hisat2, so we can use ".." to specify go one directory back then go into reads.
- Using the "-S" flag, we indicate that we want to save the alignment results in the SAM format where SAM stands for Sequencing Alignment Mapped.
hisat2 -x ../refs/22 -1 ../reads/HBR_1_R1.fq -2 ../reads/HBR_1_R2.fq -S HBR_1.sam
As HISAT2 is running the alignment, we get the alignment statistics below.
118571 (100.00%) were paired; of these:
56230 (47.42%) aligned concordantly 0 times
61769 (52.09%) aligned concordantly exactly 1 time
572 (0.48%) aligned concordantly >1 times
----
56230 pairs aligned concordantly 0 times; of these:
173 (0.31%) aligned discordantly 1 time
----
56057 pairs aligned 0 times concordantly or discordantly; of these:
112114 mates make up the pairs; of these:
112061 (99.95%) aligned 0 times
48 (0.04%) aligned exactly 1 time
5 (0.00%) aligned >1 times
52.75% overall alignment rate
Let's breakdown the alignment statistics shown above for the sample HBR_1.
The first line of the HISAT2 alignment statistics says 118571 reads (100.00%) were paired. Recall from FASTQC that read 1 and read 2 FASTQ files for HBR_1 have 118571 reads, each (Figures 1 and 2). So the first line in the HISAT2 alignment statistics is telling us that out of all the reads from read 1 and read 2 FASTQ files of HBR_1, we have 118571 pairs, which agrees with what we know. Also, this means we have 118571x2 or 237142 reads for the HBR_1 sample.
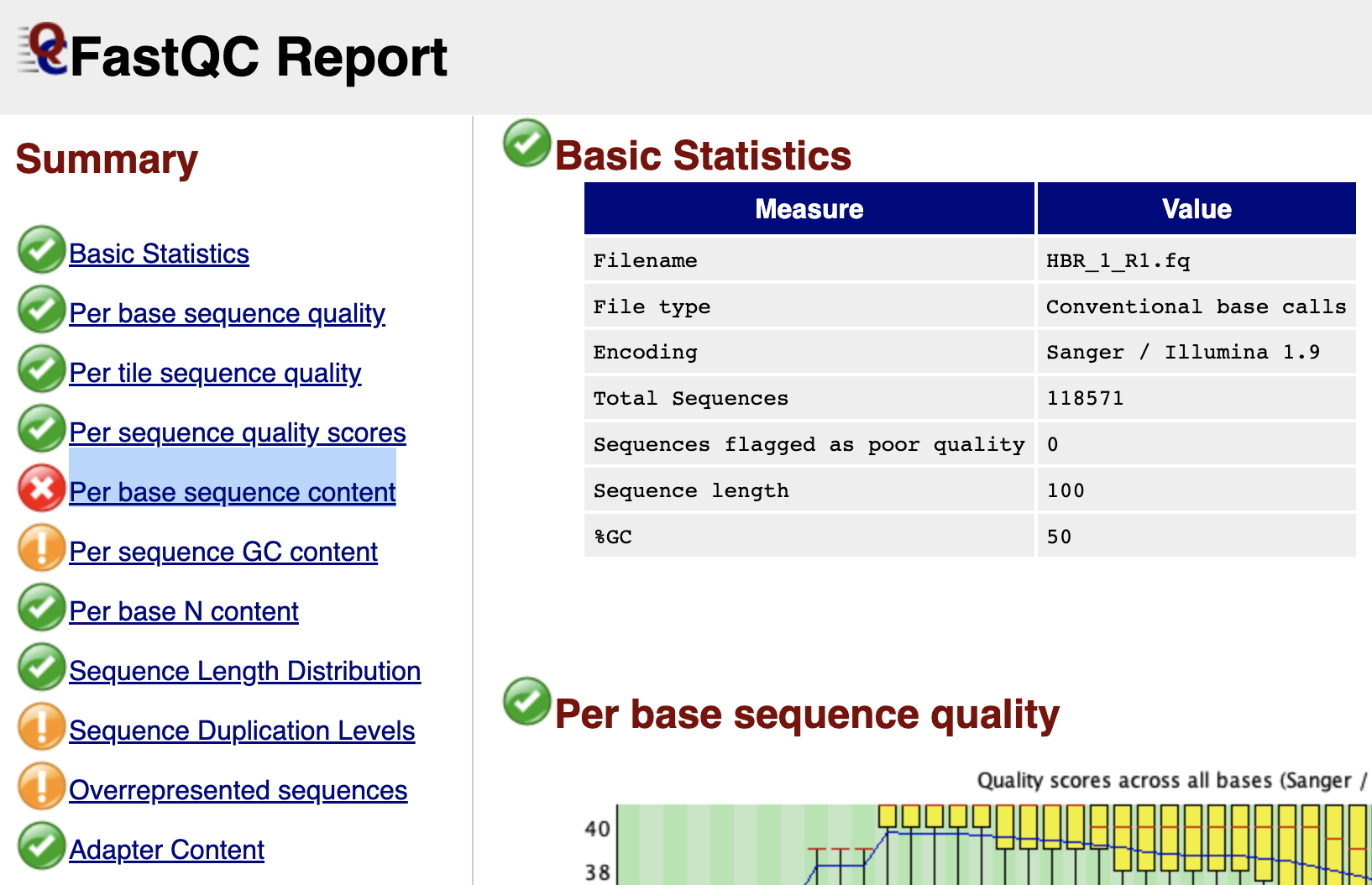
Figure 1
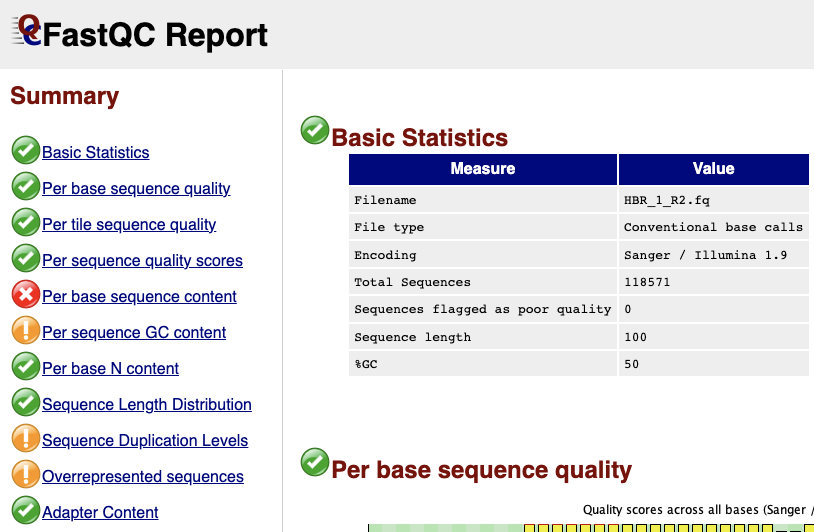
Figure 2
Following the first line of the HISAT2 alignment statistics, we will see some terminologies like concordant or discordant mapped reads. See Figure 3 for a visual explanation.
- A concordant read pair is defined as those that align in an expected manner where the reads are oriented towards one another and the distance between the outer edges is within expected ranges.
- A discordant read pair is defined as those that do not align as expected such as where the distance between the outer edges is smaller or larger than expected.
For the HBR_1 sample, we have
- 61769 pairs (123538 reads) that aligned concordantly once
- 572 pairs (1144 reads) that aligned concordantly more than once - these are mapped to multiple parts of the genome and likely cause by duplicated regions in the genome
- 173 paris (346 reads) that aligned discordantly once
- 48 reads that did align (neither concordantly or discordantly) once
- 5 reads that aligned (neither concordantly or discordantly) more than once
If we sum up the number of reads that mapped in the above break down and divide by the total number of reads in the two FASTQ files for the HBR_1 sample then we should get an overall alignment rate of 52.75%.

Figure 3: Source: Benjamin J. Raphael, Chapter 6: Structural Variation and Medical Genomics, PLOS Computational Biology, December 27, 2012
If we include the option --summary-file in the HISAT2 command, we can specify a file name to save the alignment statistcs.
Before moving further, let's use the parallel and echo commands to create a text file that contains the IDs for the HBR and UHR samples. This will allow us to align many FASTQ files at the same time.
To create the list of sample IDs for the HBR and UHR dataset we run the command below:
- parallel allows us to create the list of sample IDs all in one go (ie. multi-task rather than doing things in series)
- echo will print its arguments to the terminal (ie. echo horse will print horse in the terminal), in this case we want echo to print {1}{2}, where these can represent anything connected together by the underscore (the underscore is denoted by ). {1} denotes input 1 and {2} denotes input 2 such that input 1 is printed first and input 2 is printed second.
- the inputs in the command below are HBR and UHR, the sample groups in our dataset; "1 2 3" to denote 1, 2, or 3 replicates for each group (ie. we have samples HBR_1, HBR_2, HBR_3, UHR_1, UHR_2, UHR_3)
- one of the ways to specify input using parallel is the ":::" notation. here, after {1}_{2}, we specify the sample groups as the first input (HBR UHR) and then the replicate number (1, 2, or 3)
- we write this to a file called ids.txt, in the ~/biostar_class/hbr_uhr/reads folder, where the reads folder is one directory up from our present working directory of ~/biostar_class/hbr_uhr/snidget_hisat2 so we can denote this using ".." (one directory up) and then specify the reads directory followed by the file name
parallel echo {1}_{2} ::: HBR UHR ::: 1 2 3 > ../reads/ids.txt
cat ../reads/ids.txt
HBR_1
HBR_2
HBR_3
UHR_1
UHR_2
UHR_3
To align all of the HBR and UHR FASTQ files we can take advantage of the parallel command. Let's use the --summary-file option to store the alignment statistics. We will see why this is useful in a bit. In the command below, we
- read the sample names stored in ~/biostar_class/hbr_uhr/reads/ids.txt using cat
- send this to parallel and the hisat2 command is enclosed in double quotes within
- we use {} as a place holder for accepting the sample IDs provided by the cat command
- because we are dealing with paired-end sequencing, we append _R1 and _R2 to denote the first and second file in the pair
cat ../reads/ids.txt | parallel "hisat2 -x ../refs/22 -1 ../reads/{}_R1.fq -2 ../reads/{}_R2.fq -S {}.sam --summary-file {}_hisat2_summary.txt"
After alignment with HISAT2, let's list the contents of the hbr_uhr_hisat2 directory to see what has changed.
ls -1
HBR_1.sam
HBR_1_hisat2_summary.txt
HBR_2.sam
HBR_2_hisat2_summary.txt
HBR_3.sam
HBR_3_hisat2_summary.txt
UHR_1.sam
UHR_1_hisat2_summary.txt
UHR_2.sam
UHR_2_hisat2_summary.txt
UHR_3.sam
UHR_3_hisat2_summary.txt
If we print the alignment summary file for HBR_1 (HBR_1_hisat2_summary.txt) then we should see the same statistics that we saw earlier.
cat HBR_1_hisat2_summary.txt
Recall from Lesson 11 that we can include summaries from post alignment steps into MultiQC reports. This is why we create the summary files for the HISAT2 alignments for the HBR and UHR data. So let's run MultiQC again to generate a report that includes the HISAT2 alignment statistics. Make sure that we change the ~/biostar_class/hbr_uhr/ folder of our home directory and then construct the multiqc command below, where we specify the output filename using --filename. The output will be written in the QC directory. We use "." to tell multiqc to search ~/biostar_class/hbr_uhr for any QC or log files and it will search not only the present working directory but also sub-directories.
cd ~/biostar_class/hbr_uhr
multiqc --filename QC/multiqc_hbr_uhr_with_hisat2 .
Note that multiqc is now adding the alignment statistics in the report.
/// MultiQC 🔍 | v1.13
| multiqc | Search path : /home/joe/biostar_class/hbr_uhr
| searching | ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 82/82
| snippy | Found 1 reports
| bargraph | Tried to make bar plot, but had no data: snippy_variants
| bowtie2 | Found 6 reports
| fastqc | Found 14 reports
| multiqc | Compressing plot data
| multiqc | Report : qc/multiqc_with_hisat2.html
| multiqc | Data : qc/multiqc_with_hisat2_data
| multiqc | MultiQC complete
We can copy multiqc_hbr_uhr_with_hisat2 to ~/public and then take a look at this file.
cp QC/multiqc_hbr_uhr_with_hisat2.html ~/public
After multiqc_hbr_uhr_with_hisat2.html has been copied to the ~/public directory, go back to the GOLD landing page (you can access it by clicking on the Class html links).
At the GOLD landing page, scroll down the student table until you see your name and click the tab labeled File that is associated with your name.
You will then be taken to a page that where you access the files in your ~/public directory. You can either right click to download the files or view in a separate browser tab in the case of html files.

In the navigation pane of the multiqc report, we now see a link to the hisat2 alignment statistics (Figure 4).
Figure 4
In the general statistics table, we now have a column indicating the overall alignment rate for each sample (Figure 5).
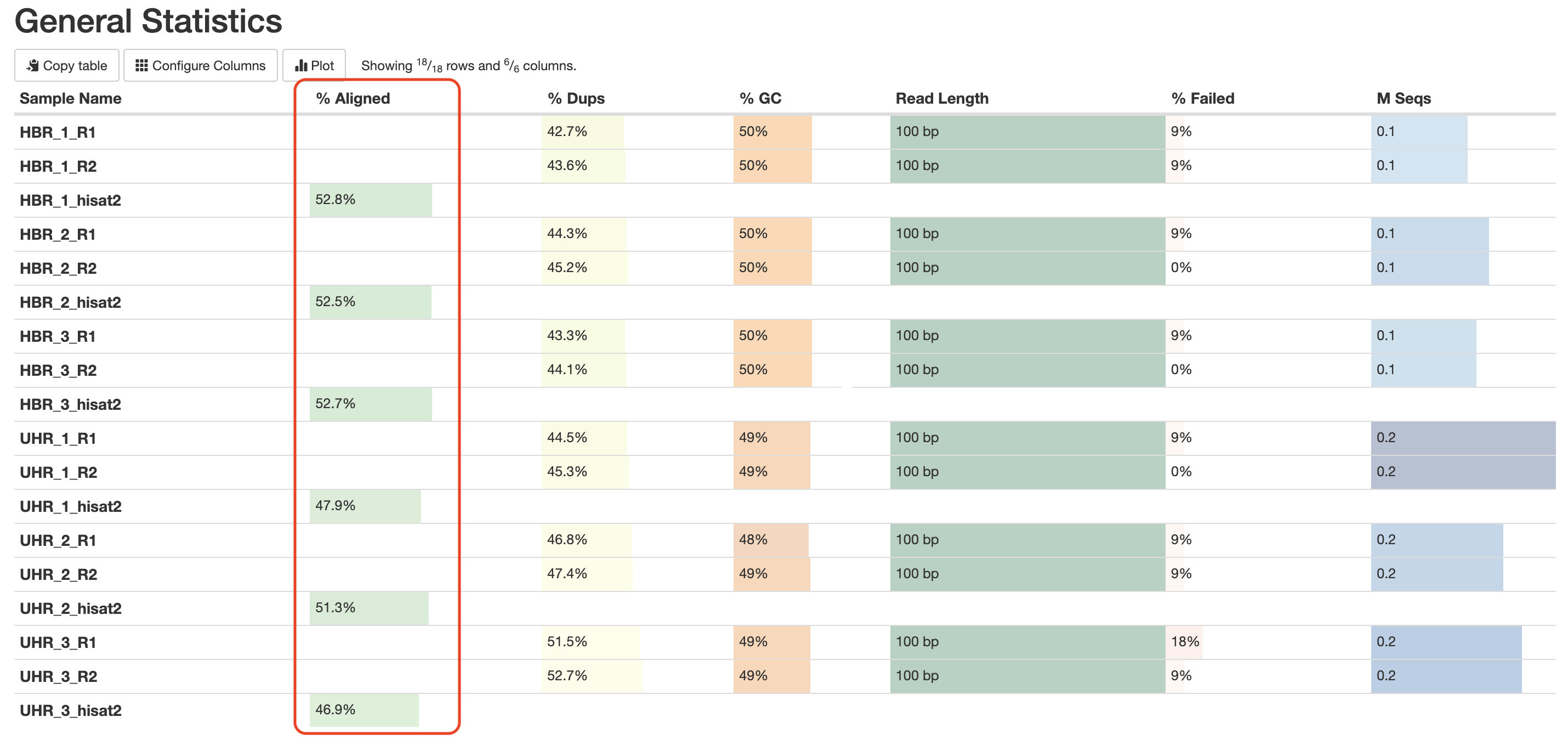
Figure 5
We also have a plot (Figure 6) that tells us
- How many pair of reads mapped uniquely (ie. only once)
- How many pair of reads mapped concordantly to more than one location
- How many pair of reads mapped uniquely but discordantly
- How many pair of reads did not align
- How many pair of reads where one of the pair mapped uniquely
- How many pair of reads where one of the pairs were multimapped
This information is the same that we see in the alignment statistics generated by HISAT2, except now we have combined it with our pre-alignment QC reports and this provides a nice way for us to keep the logs and summary of our analysis in one place so we can share with colleagues and collaborators.
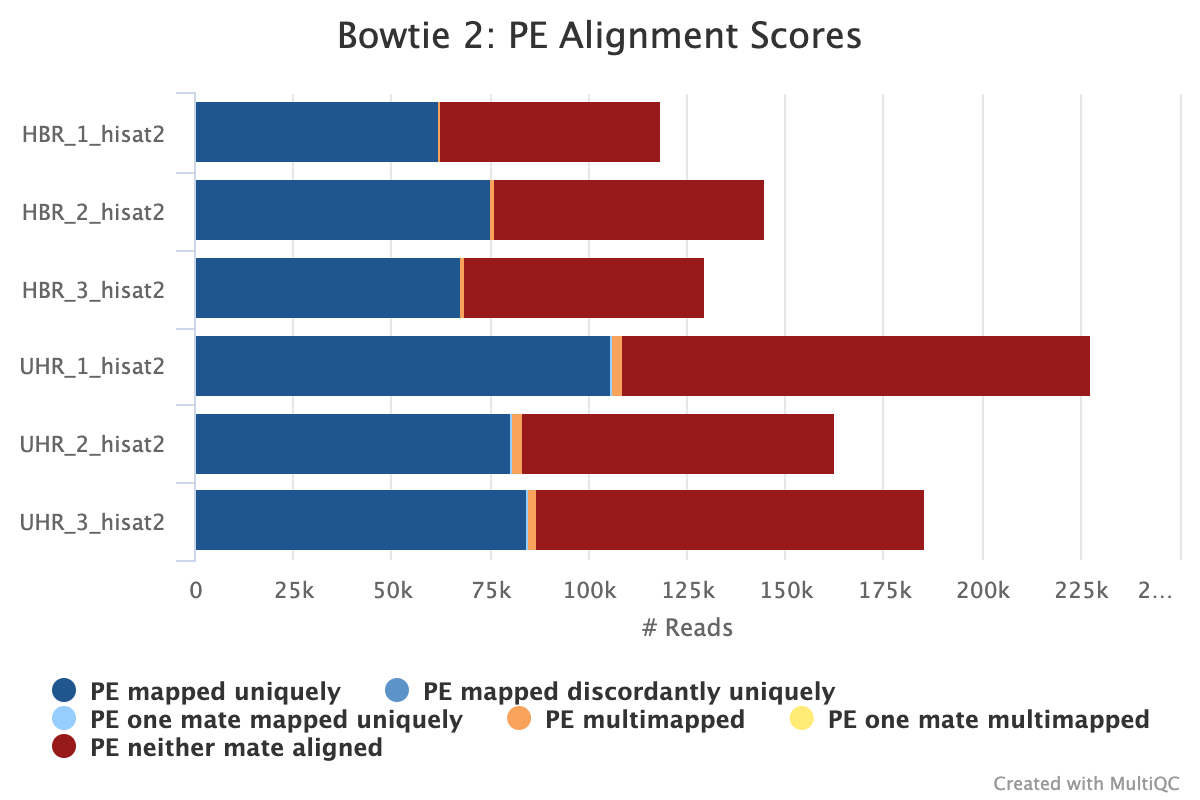
Figure 6
If we click on the bar for one of the samples, a dialogue box with alignment statistics appears and we can then see the numbers.

Figure 7
Working with SAM file alignment outputs
Change in the ~/biostar_class/hbr_uhr/hbr_uhr_hisat2 folder for this portion of the class.
cd ~/biostar_class/hbr_uhr/hbr_uhr_hisat2
Now that we have our SAM files generated for the HBR and UHR dataset. For your reference, information on the SAM file can be found here. A SAM file is tab delimited, so we can opened using Excel. I am going to copy HBR_1.sam to my ~/public folder so I can open this locally on my Excel to go over this SAM file (try to just watch what I do here).
SAM files always start off with metadata information and these lines start with @.
- @HD includes
- SAM file format information (in this case version 1.0 as indicated by VN:1.0)
- Whether the alignment file has been sorted (in this case no as indicated by SO:unsorted)
- @SQ provides reference information
- SN denotes reference sequence name, which is chr22
- LN is the reference length in bases, which is 50818468 as we found in Lesson 9 using seqkit stats
- @PG provides information about the program we used to generate the alignment
- ID is the program record identifier
- PN is the program name (HISAT2 in our case version 2.2.1 as indicated next to VN)
- CL is the command line call used to generate the alignment
After the metadata information, each SAM file contains 11 fields.
- QNAME or query template name, which is essentially the sequencing read that we want mapped onto a reference genome. If we grep the first sequence in the HBR_1.sam, then we should retrieve one of the reads in the original FASTQ files (try it on your own).
- The second column is a FLAG that tells us a bit about the mapping. Here is a good tool to help interpret these FLAGS. These FLAGS can inform us of things like pair end read alignment orientation.
- Column three contains the name of our reference genome.
- Column four tells us the left most genomic coordinate where the sequencing read maps.
- The mapping quality (MAPQ) is provided in the fifth column (the higher the number, the more confident we are of the map); a value of 255 in this column means that the mapping quality is not available.
- Column six presents the CIGAR string, which tells us information about the match/mismatch, insertion/deletion, etc. of the alignment.
- Column seven is the reference sequence name of the primary alignment of the NEXT read in the template. We will see an "=" if this name is the same as the current (which we would expect for paired reads)
- The alignment position of the next read in the template is provided in column 8. When this is not available, the value is set to 0.
- Column nine provides the template length (TLEN), which tells us how many bases of the reference genome does the sequencing read span.
- The tenth column is just the sequencing read (some are written as the reverse complement so be cautious. The FLAGS in column two will tell us whether the sequence is reverse complemented).
- The eleventh column is the Phred quality scores of the sequencing read.
- For definition of optional fields, see https://samtools.github.io/hts-specs/.
The SAM file format is human readable so we will need to convert it to a machine readable format (Binary Alignment Map or BAM) before we can visualize alignment results using IGV and perform other downstream processes like obtaining read counts. To convert between SAM and BAM, we can use an application called SAMTOOLS.
If we type samtools on the command line, we can see that this application has lots of features. For an alignment file to be of use, we will need to sort it by genomic position using the sort feature of SAMTOOLS. So we type the following command where the -o flag prompts us to enter the output file name (in this case it will be a BAM file). The last argument is the input SAM file, which is the SAM file that we want to sort.
samtools sort -o HBR_1.bam HBR_1.sam
Now that we have our BAM file for HBR_1 generated, we need to index it. Similar to the idea of indexing a reference genome, indexing the BAM file will allow the program that uses it to more efficiently search through it. For this we will use samtools index, where the -b flag tells SAMTOOLS to create the index from a BAM file. We include the extension ".bai" in the output file.
samtools index -b HBR_1.bam HBR_1.bam.bai
The sorting and indexing of our BAM files are perhaps the two necessary steps that allows us to visualize and move forward with our analysis. The above shows how to sort and index one BAM file at a time. Below, let's use the parallel command to take care of these tasks for all of our alignment outputs at one go.
Similar to how we constructed the hisat2 alignment, we use cat to send the sample IDs to parallel. The samtools command construct is enclosed in double quotes and {} acts as a place holder for accepting the sample IDs provided by the cat command.
cat ../reads/ids.txt | parallel "samtools sort -o {}.bam {}.sam"
cat ../reads/ids.txt | parallel "samtools index -b {}.bam {}.bam.bai"
Alignment of HBR and UHR raw sequencing data with Bowtie2
For RNA sequencing studies, we need to use a splice aware aligner to account for reads that map across exons. Bowtie2 is a commonly used aligner for DNA sequencing and is not splice aware. Let's align the HBR and UHR data to human chromosome 22 using Bowtie2 so we can see how the alignment results differ from HISAT2 when we visualize alignment results in Lesson 14.
To run Bowtie2, we will need to create a Bowtie2 specific index for chromosome 22, so change into the ~/biostar_class/hbr_uhr/refs folder.
cd ~/biostar_class/hbr_uhr/refs
Then, we use bowtie2-build using 22.fa as input (like we did with HISAT2) and assign to the index the base name of 22.
bowtie2-build 22.fa 22
Listing the contents of our biostar_class/hbr_uhr folder, we see the Bowtie2 indices for chromosome 22 has been created and these have extension "*.bt2"
ls -1
22.1.bt2
22.1.ht2
22.2.bt2
22.2.ht2
22.3.bt2
22.3.ht2
22.4.bt2
22.4.ht2
22.5.ht2
22.6.ht2
22.7.ht2
22.8.ht2
22.fa
22.gtf
22.rev.1.bt2
22.rev.2.bt2
ERCC92.fa
ERCC92.gtf
Before running Bowtie2 alignment, creates a folder called hbr_uhr_bowtie2 in the ~/biostar_class/hbr_uhr directory and change into.
cd ~/biostar_class/hbr_uhr
mkdir hbr_uhr_bowtie2
cd hbr_uhr_bowtie2
The construct for the command to alignment using Bowtie2 is similar to Hisat2, with the exception that we append "_bowtie2" to the output so that we know that it is from a Bowtie2 alignment. HISAT2 is actually based on Bowtie2 http://daehwankimlab.github.io/hisat2/manual/.
In the command below
- read the sample names stored in ~/biostar_class/hbr_uhr/reads/ids.txt
- send this to parallel and the bowtie2 command is enclosed in double quotes within
- we use {} as a place holder for accepting the sample IDs provided by the cat command
- because we are dealing with paired-end sequencing, we append _R1 and _R2 to denote the first and second file in the pair
cat ../reads/ids.txt | parallel "bowtie2 -x ../refs/22 -1 ../reads/{}_R1.fq -2 ../reads/{}_R2.fq -S {}_bowtie2.sam"
And now we will sort the Bowtie2 SAM files and convert to BAM.
In the commands below, we use cat to send the sample IDs to parallel. The samtools command construct is enclosed in double quotes and {} acts as a place holder for accepting the sample IDs provided by the cat command.
cat ../reads/ids.txt | parallel "samtools sort -o {}_bowtie2.bam {}_bowtie2.sam"
Finally, we will index the the Bowtie2 alignment results.
cat ../reads/ids.txt | parallel "samtools index -b {}_bowtie2.bam {}_bowtie2.bam.bai"
MultiQC reports with pre-alignment QC and post-alignment statistics
HBR and UHR MultiQC with pre- and post- alignment statistics