Alignment
RNASeq Mapping Challenges
The majority of mRNA derived from eukaryotes is the result of splicing together discontinuous exons, and this creates specific challenges for the alignment of RNASEQ data.
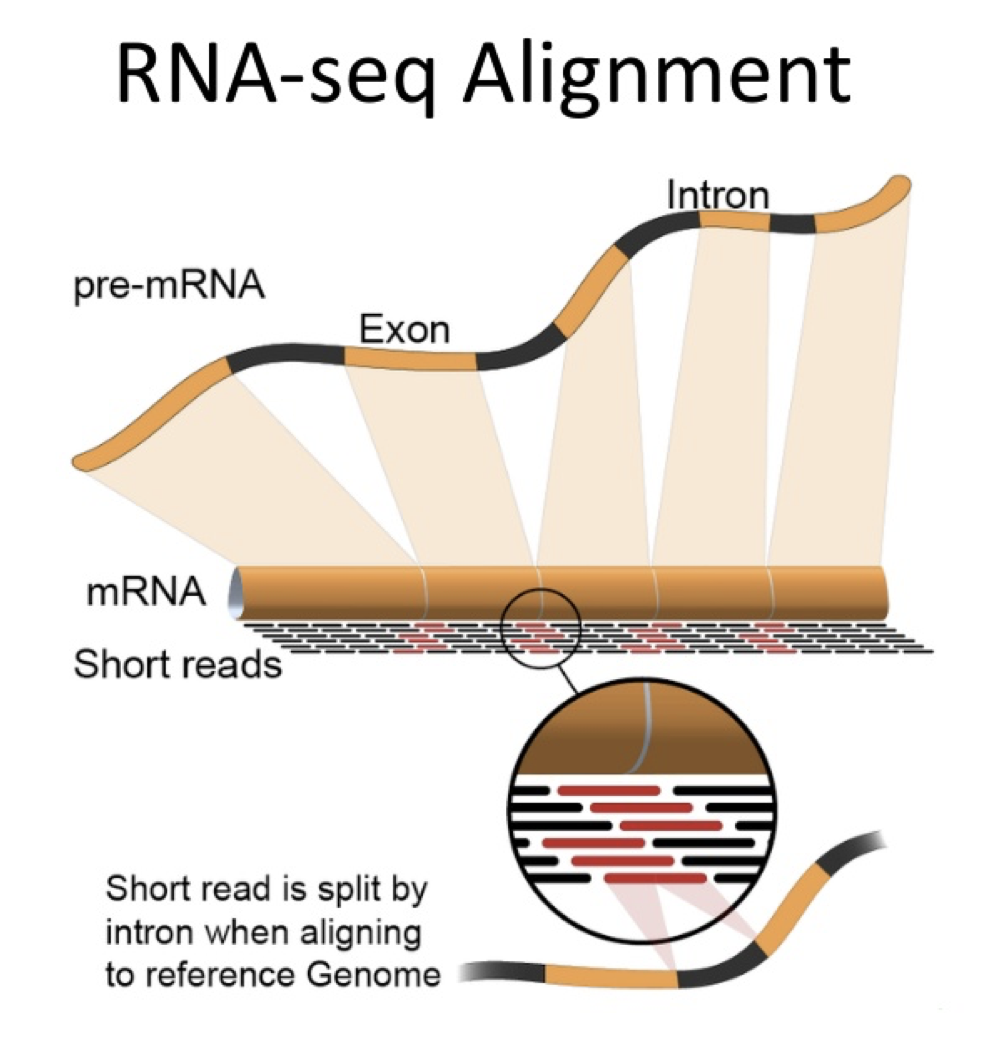
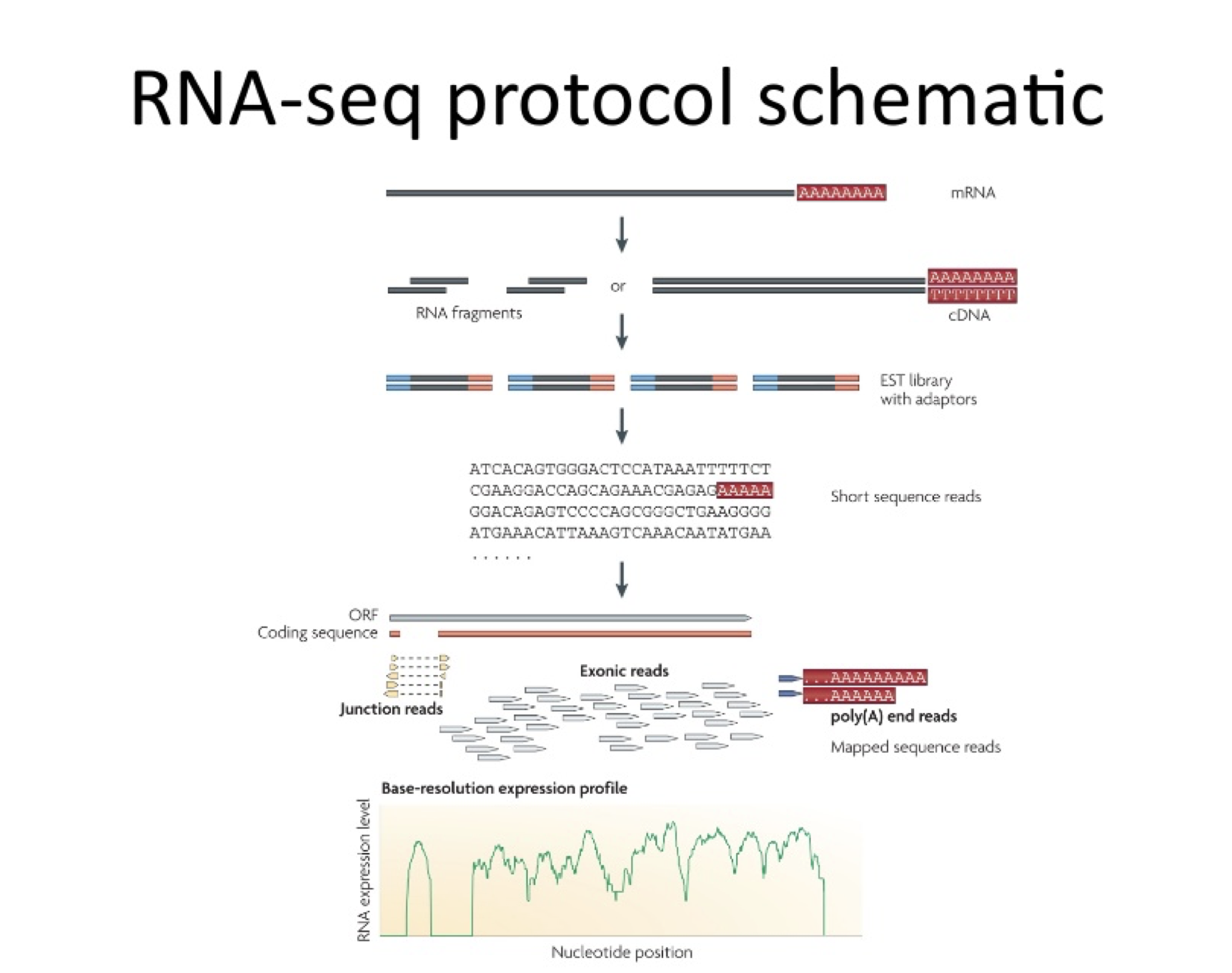
Mapping Challenges
- Reads not perfect
- Duplicate molecules (PCR artifacts skew quantitation)
- Multimapped reads - Some regions of the genome are thus classified as unmappable
- Aligners try very hard to align all reads, therefore fewest artifacts occur when all possible genomic locations are provides (genome over transcriptome)
RNASeq Mapping Solutions
There are a number of specific solutions that have been devised to address the issues created by attempting to map mRNA to DNA genomes. Each of these has its advantages and disadvantages.
-
Align against the transcriptome
- Many/All transcriptomes are incomplete
- Can only measure known genes
- Won’t detect non-coding RNAs
- Can’t look at splicing variants
- Can’t detect fusion genes or structure variants
-
De novo assembly of RNASeq reads
- Largely used for uncharacterized genomes
-
Align against the genome using a splice-aware aligner
- Most versatile solution
-
Pseudo-Aligner - quasi mappers (Salmon and Kalisto)
- New class of programs - blazingly fast
- Map to transcriptome (not genome) and does quantitation
- Surprisingly accurate except for very low abundance signals
- With bootstrapping can give confidence values
The complexity of the problem of accurately mapping millions of reads against large genomes can be appreciated by looking at a time line of the development of different mapping programs.
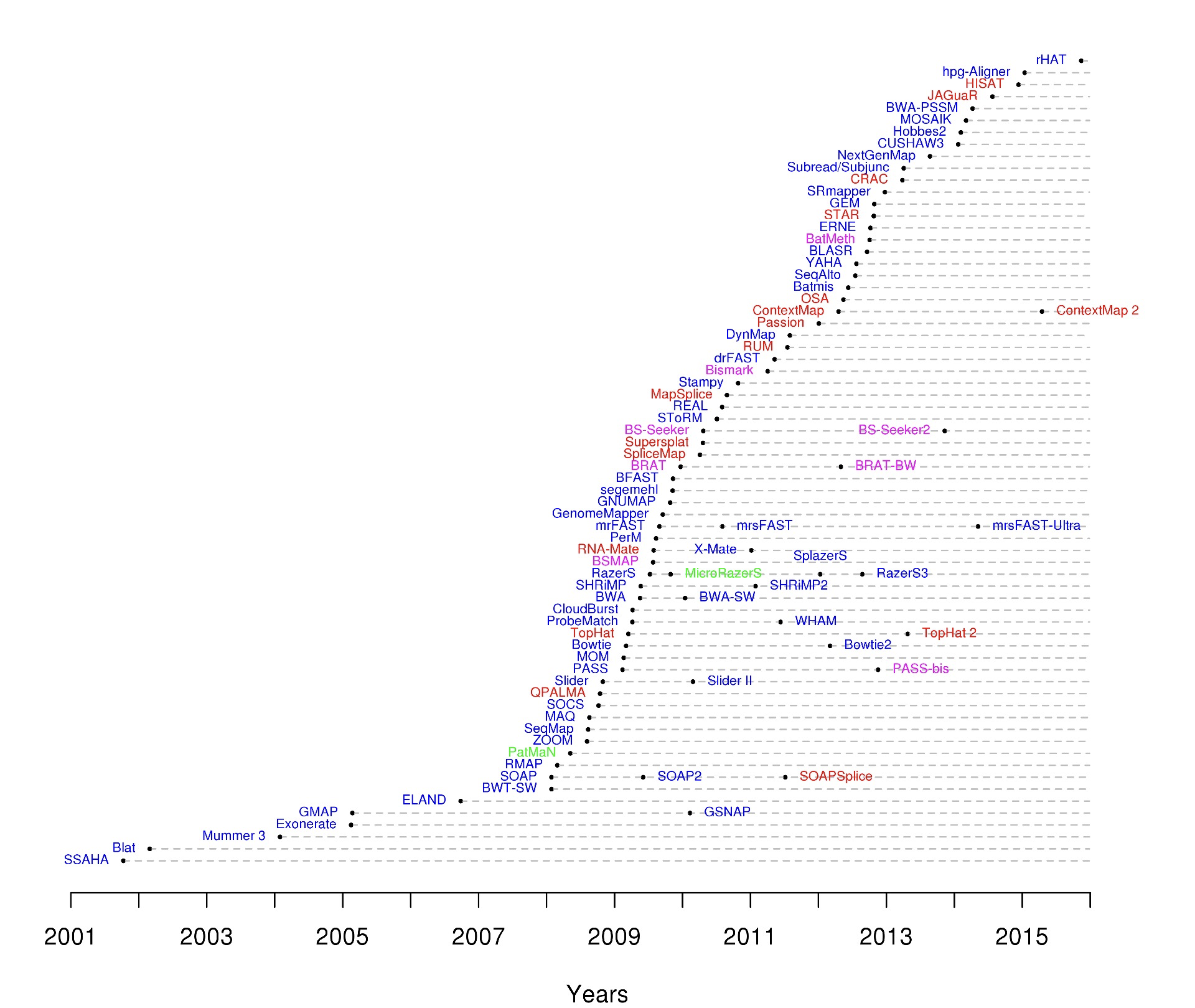
Common Aligners
Most alignment algorithms rely on the construction of auxiliary data structures, called indices, which are made for the sequence reads, the reference genome sequence, or both. Mapping algorithms can largely be grouped into two categories based on properties of their indices: algorithms based on hash tables, and algorithms based on the Burrows-Wheeler transform
- Bowtie2 BWA/BWA-mem STAR
- HISAT
- HISAT2
- TopHat
- TopHat2
Tools for mapping high-throughput sequencing data Nuno A. Fonseca Johan Rung Alvis Brazma John C. Marioni Author Notes Bioinformatics, Volume 28, Issue 24, 1 December 2012, Pages 3169–3177, https://doi.org/10.1093/bioinformatics/bts605
To Align or not to Align
Aligners typically align against the entire genome and provide a output where the results can be visibly inspected (bam file via IGV). The must be used for detecting novel genes/transcripts. Quantitation of aligned reads to specific genes is typically done by separate program
PseudoAligners assign reads to the most appropriate transcript... can’t find novel genes/transcripts or other anomalies. Generally much faster than aligner and are likely more accurate (Recent improvements in salmon have increased its accuracy, at the expense of being somewhat slower than the original)
Typical Questions about alignment
- What is the best aligner to use?
- What Genome version should I use?
- What Genome annotation should I use?
Answers
- STAR - (Salmon or Kallisto) - subjective
- Depends ! most recent or best annotated
- GeneCode with caveats - know what is being annotated and what is not and how it effects your results
Questions not asked
- What parameters should I use?
Answers
Most programs have lots of optional parameters that can tweak the results, but most are set to defaults that should work in most common situations. (Don’t touch what you don’t understand - especially if it gets you, your favorite answer)
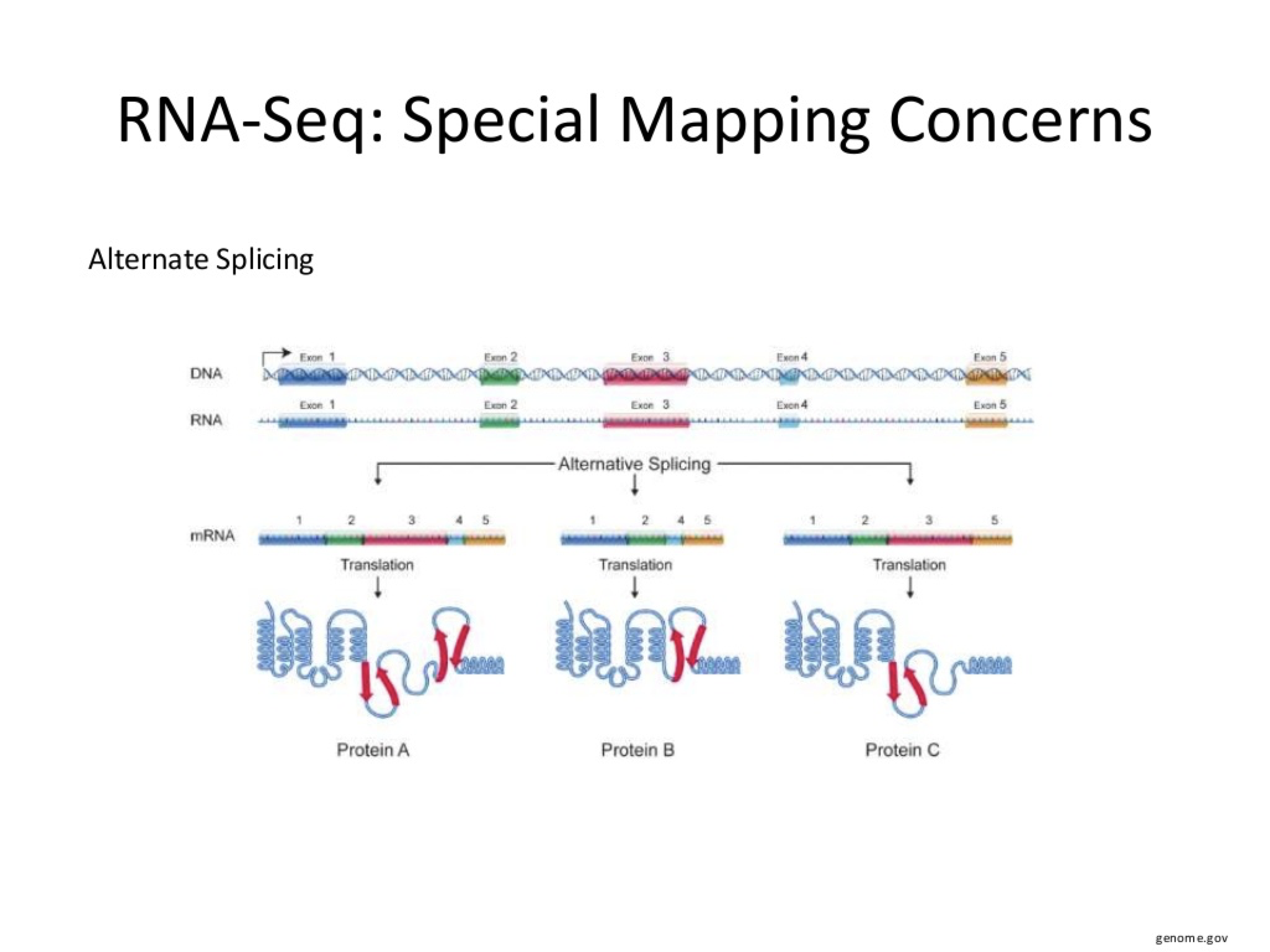
Special Consideration for Alternate Splicing Events
To add to the mRNA mapping problem is the existance of alternate splicing events. Attempting to identify alternate splicing in RNASEQ data is not something for the novice to attempt! .... get professional help
Post Alignment QC Programs
RSeQC package provides a number of useful modules that can comprehensively evaluate high throughput sequence data especially RNA-seq data. “Basic modules” quickly inspect sequence quality, nucleotide composition bias, PCR bias and GC bias, while “RNA-seq specific modules” investigate sequencing saturation status of both splicing junction detection and expression estimation, mapped reads clipping profile, mapped reads distribution, coverage uniformity over gene body, reproducibility, strand specificity and splice junction annotation.
MultiQC is a modular tool to aggregate results from bioinformatics analyses across many samples into a single report.
Picard Tools - RNAseqMetrics is a module that produces metrics about the alignment of RNA-seq reads within a SAM file to genes
RSeQC example of plot types
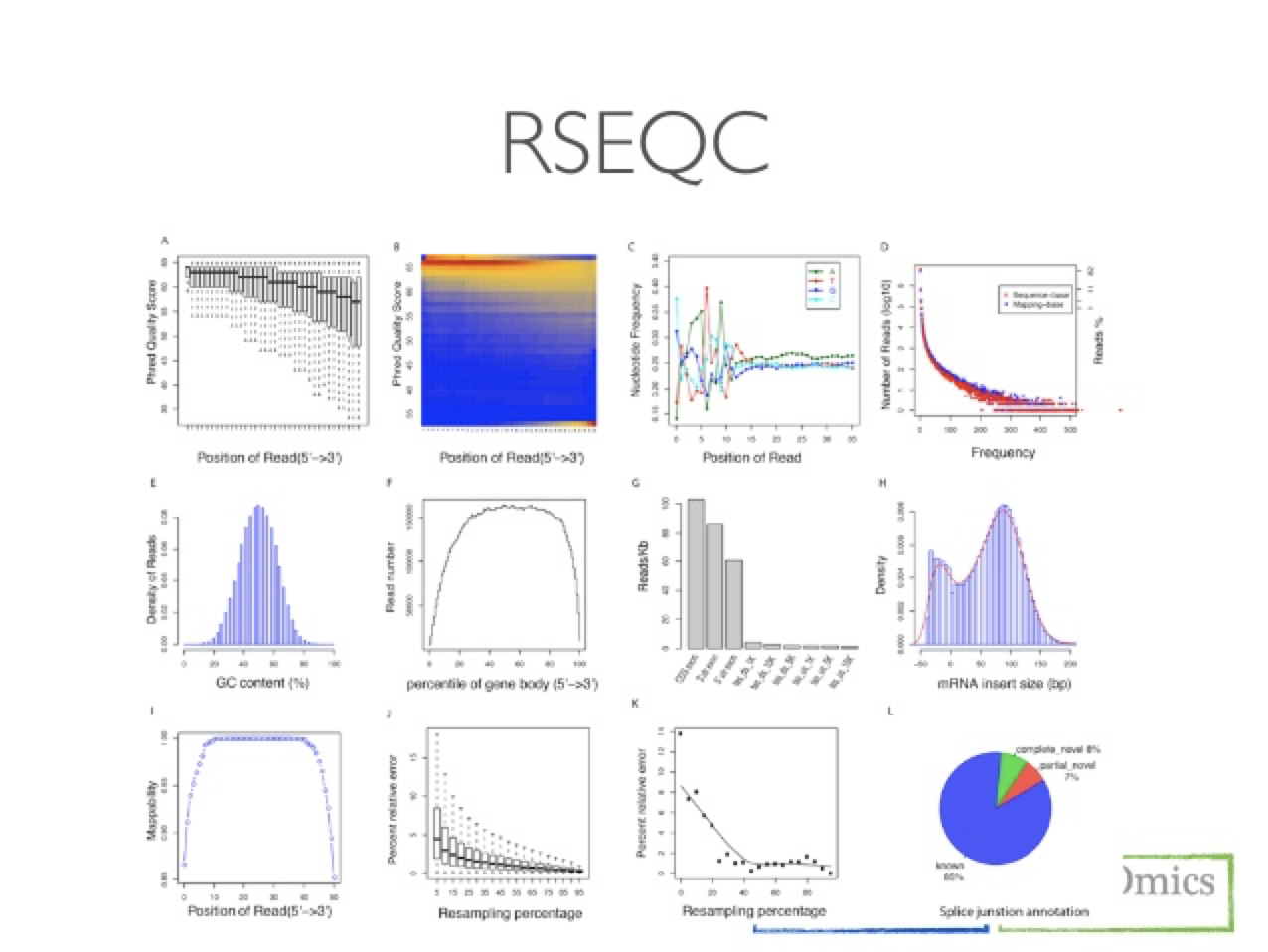
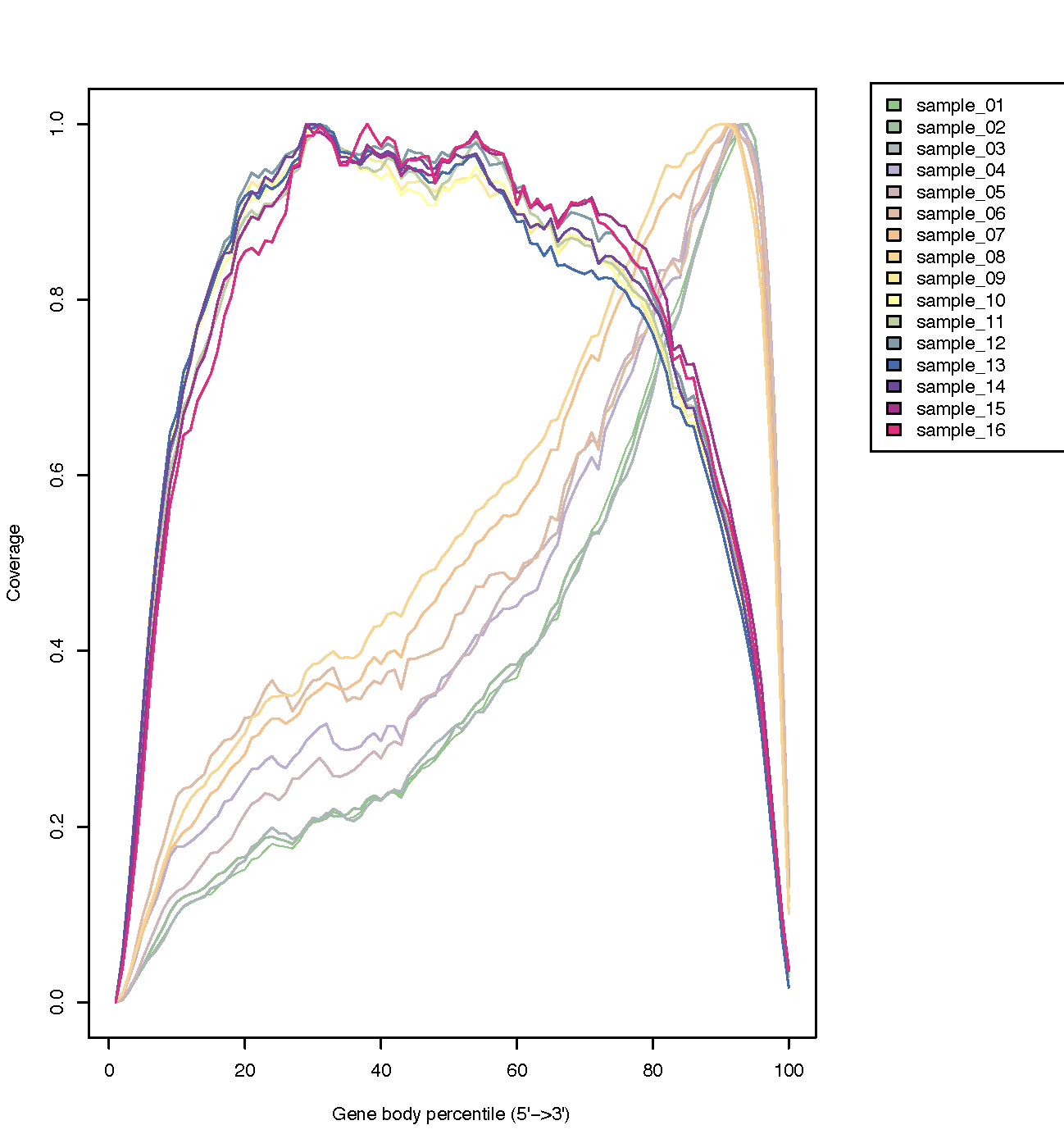
Post Alignment Cleanup Programs
Picard is a set of command line tools for manipulating high- throughput sequencing (HTS) data and formats such as SAM/ BAM/CRAM and VCF. (mark pcr duplicates)
Samtools provide various utilities for manipulating alignments in the SAM/BAM format, including sorting, merging, indexing and generating alignments in a per-position format.
BamTools is a command-line toolkit for reading, writing, and manipulating BAM (genome alignment) files.