Lesson 11 Practice
Objectives
In this lesson, we learned to
- merge multiple FASTQC reports into one
- perform data cleanup (quality and adapter trimming) to prepare our sequencing reads for downstream analysis. Here, we will put what we learned to practice.
Merging Golden Snidget FASTQC reports into one
Before getting started, we should change into the ~/biostar_class/snidget/QC directory where the Golden Snidget sequencing data and FASTQC reports were saved. How do we do this?
Solution
cd ~/biostar_class/snidget/QC
How do we merge the FASTQC results from the Golden Snidget dataset into one?
Solution
Since we are in the snidget folder, which contains our FASTQC results, we can use "." to denote "here in this folder" because MultiQC will look for output logs in the specify folder.
multiqc --filename multiqc_report_snidget .
Next copy the MultiQC output to the public directory. Do you remember how to do this?
Solution
cp multiqc_report_snidget.html ~/public/multiqc_report_snidget.html
Can you configure the Golden Snidget MultiQC output's General Statistics table to show the percentage of modules that failed?
In the General Statistics table of the Golden Snidget MultiQC report, can you assign different colors to distinguish the FASTQ files for the BORED and EXCITED groups?
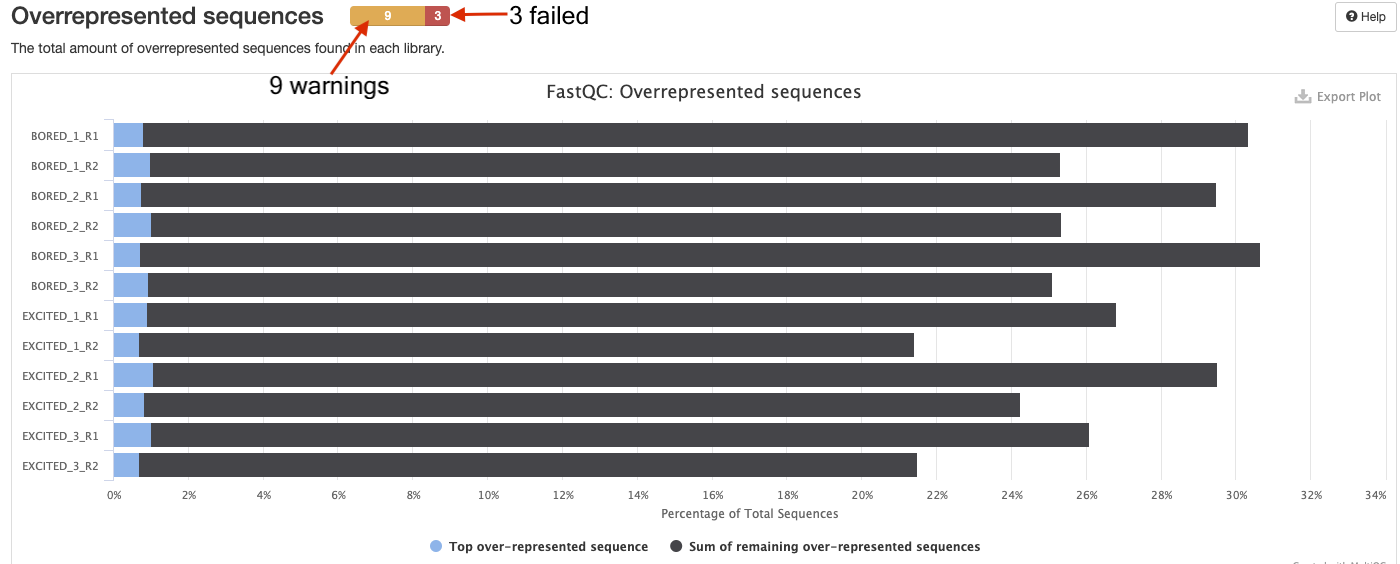
In the overrepresented sequences plot, how many samples have warnings and how many failed?
Solutions
Golden Snidget FASTQC files MultiQC results.
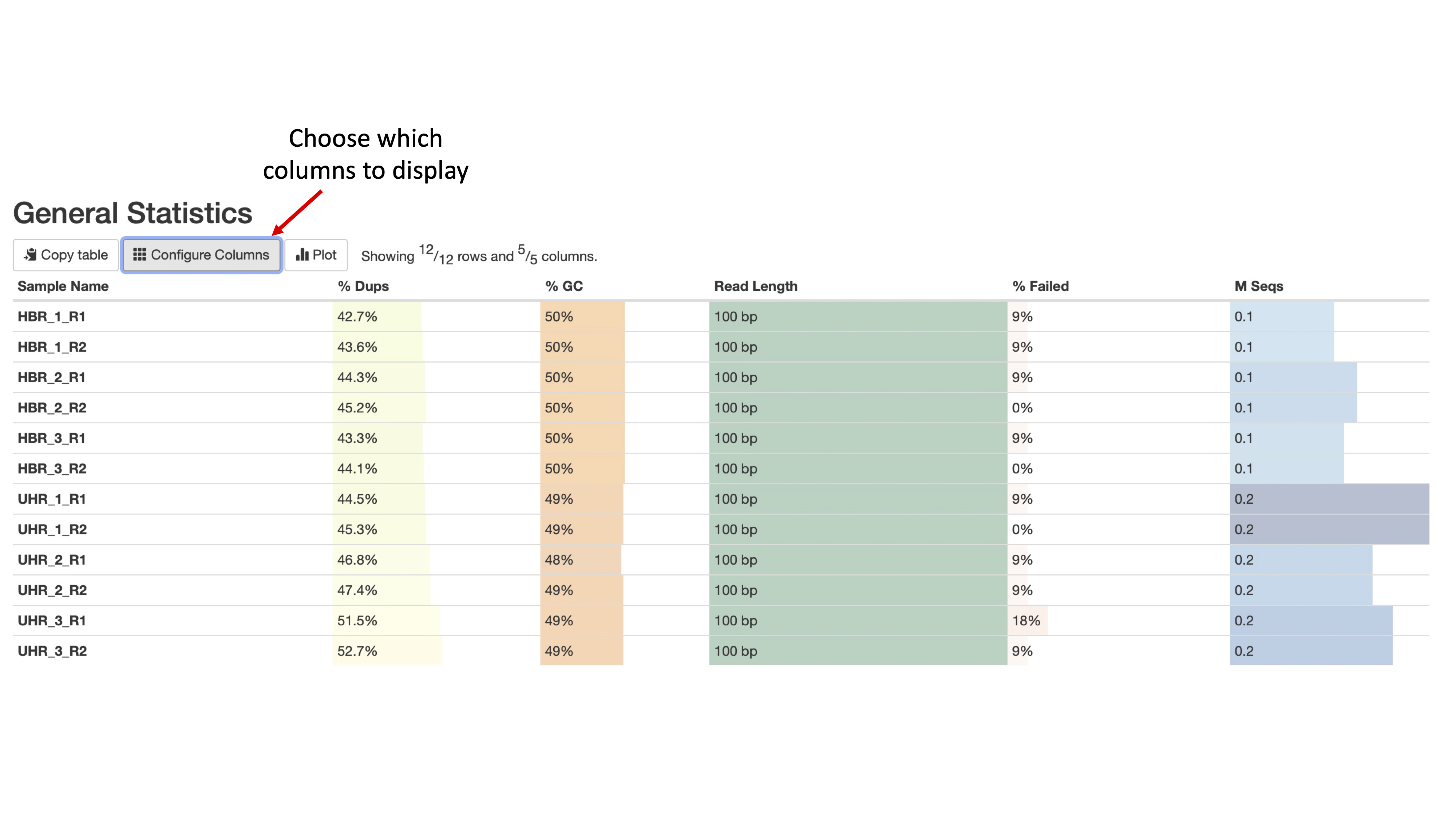
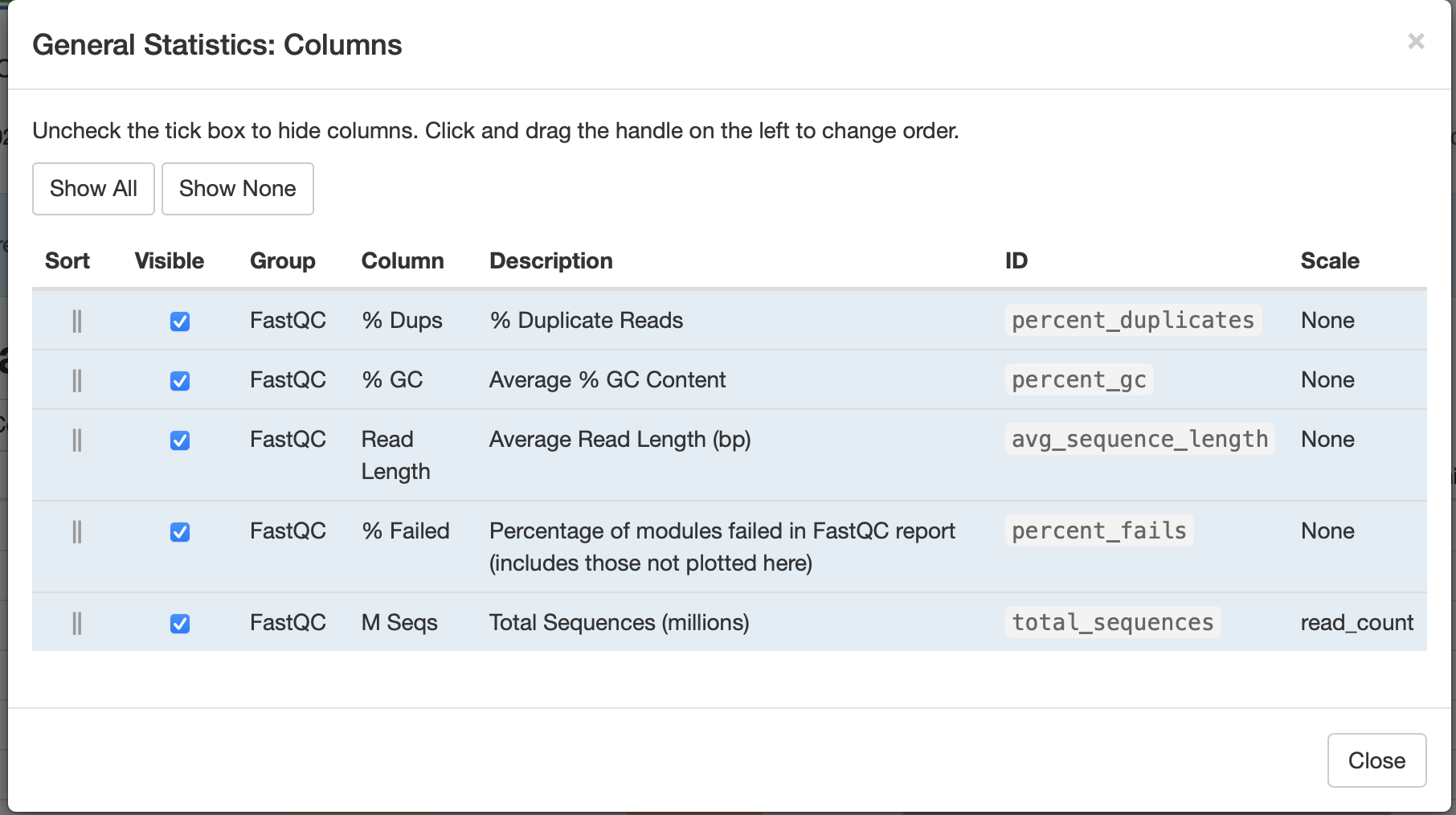

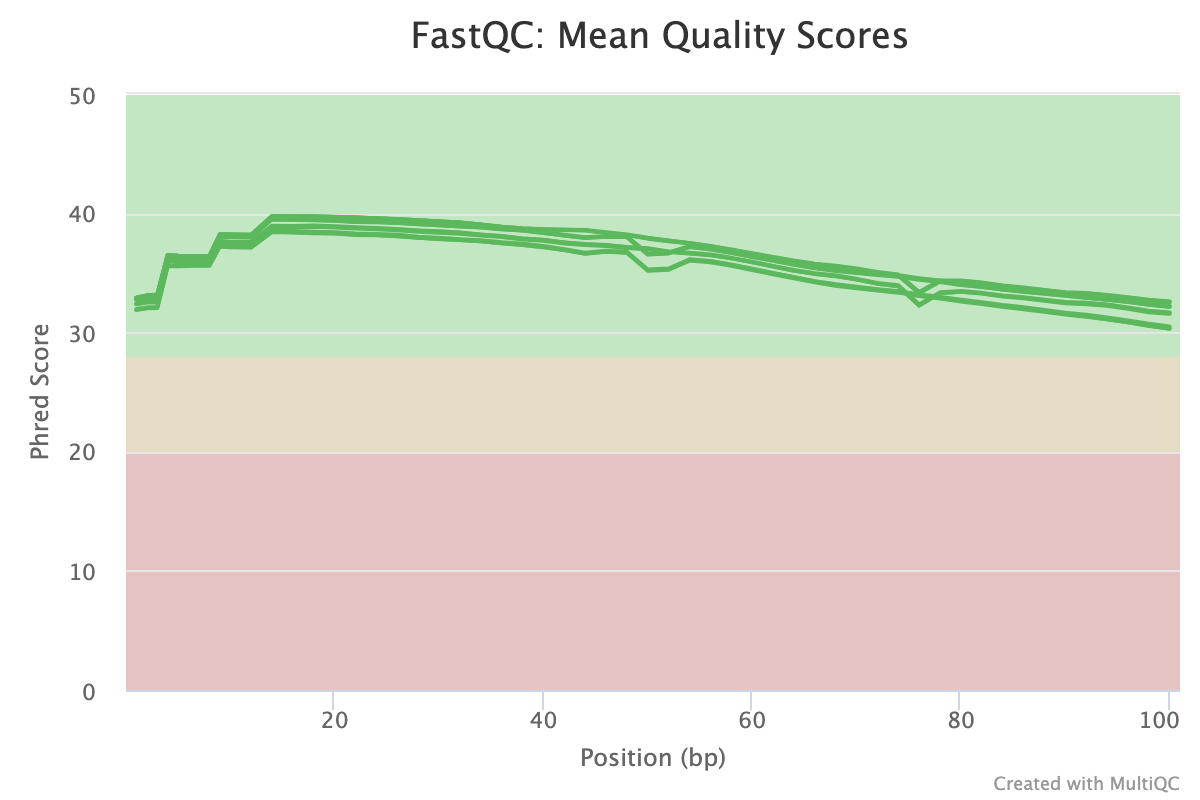






Quality and adapter trimming
Let's go back to the biostar_class directory and create a folder called practice_trimming for this exercise. How do we do this?
Solution
This depends on where you are currently (ie. your present working directory is). From there go back to the biostar_class folder.
cd ~/biostar_class
mkdir practice_trimming
After the "practice_trimming" directory has been created, change into this directory. How do we do this?
Solution
cd practice_trimming
Next, download a FASTQ file from NCBI/SRA to practice trimming with.
fastq-dump --split-files -X 10000 SRR1553606
Once the download is complete the message below will appear.
Read 10000 spots for SRR1553606
Written 10000 spots for SRR1553606
How many FASTQ files were downloaded? And from the file names, is this from paired or single end sequencing.
Solution
ls
Two FASTQ files were downloaded and this is paired end sequencing.
Let's run FASTQC for the these files. Do you recall how to do this?
Solution
fastqc SRR1553606_*.fastq
Copy the FASTQC outputs (html files) to the public directory.
Solution
cp SRR1553606_*_fastqc.html ~/public
How is the quality and are there adapter contamination for the FASTQ files in SRR1553606? If yes, can we trim away the adapters and poor quality reads? FYI, for this exercise our adapter sequence is below (can we create an input file called nextera_adapter.fa with the adapter sequence?).
>nextera
CTGTCTCTTATACACATCTCCGAGCCCACGAGAC
Solution
The answer is the quality for both FASTQ files is not great and we can remove the poor quality reads and the adapters.
nano nextera_adapter.fa
Copy and paste the adapter sequence into nano, hit control x and save to exit.
>nextera
CTGTCTCTTATACACATCTCCGAGCCCACGAGAC
trimmomatic PE SRR1553606_1.fastq SRR1553606_2.fastq SRR1553606_trimmed_1.fastq SRR1553606_trimmed_1_unpaired.fastq SRR1553606_trimmed_2.fastq SRR1553606_trimmed_2_unpaired.fastq SLIDINGWINDOW:4:30 ILLUMINACLIP:nextera_adapter.fa:2:30:5 MINLEN:50
Run FASTQC on the trimmed output. Any improvements?
Solution
fastqc SRR1553606_trimmed_1.fastq SRR1553606_trimmed_2.fastq
What is another tool that we can use to perform quality and adapter trimming on FASTQ files?
Solution
BBDuk
bbduk.sh in=SRR1553606_1.fastq in2=SRR1553606_2.fastq out=SRR1553606_bbduk_1.fastq out2=SRR1553606_bbduk_2.fastq qtrim=r overwrite=true trimq=30 ref=nextera_adapter.fa ktrim=r k=16 mink=10 hdist=1 tpe minlen=50